Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Untitled
Vol 445 8 February 2007 doi:10.1038/nature05529
Senescence and tumour clearance is triggered by p53restoration in murine liver carcinomas
Wen Xue1*, Lars Zender1*, Cornelius Miething1, Ross A. Dickins1,2, Eva Hernando3, Valery Krizhanovsky1,Carlos Cordon-Cardo3 & Scott W. Lowe1,2
Although cancer arises from a combination of mutations in onco-
Animals with advanced tumours were treated with Dox to re-
genes and tumour suppressor genes, the extent to which tumour
establish p53 expression (Fig. 1b). Shortly after Dox administration,
suppressor gene loss is required for maintaining established
the p53 microRNA (miRNA) was shut off and p53 expression
tumours is poorly understood. p53 is an important tumour sup-
increased (Supplementary Fig. 1). Although tumours in untreated
pressor that acts to restrict proliferation in response to DNA
mice rapidly progressed, those in Dox-treated animals began to
damage or deregulation of mitogenic oncogenes, by leading to
involute and became nearly undetectable within 12 days (Fig. 1b).
the induction of various cell cycle checkpoints, apoptosis or cel-
Similar results were observed in a subcutaneous setting, where
lular senescence1,2. Consequently, p53 mutations increase cell pro-
tumours could be accurately monitored using calliper measurements
liferation and survival, and in some settings promote genomic
(Fig. 1c, left panel). Importantly, ras-induced liver carcinomas pro-
instability and resistance to certain chemotherapies3. To deter-
duced using a constitutive p53 shRNA grew similarly irrespective of
mine the consequences of reactivating the p53 pathway in
Dox treatment (Fig. 1c, right), indicating that tumour regression was
tumours, we used RNA interference (RNAi) to conditionally regu-
not due to Dox toxicity. Such regressions also occurred when p53 was
late endogenous p53 expression in a mosaic mouse model of liver
reactivated in tumours co-expressing a constitutively activated Akt
carcinoma4,5. We show that even brief reactivation of endogenous
or an endogenous oncogenic K-ras allele and the conditional p53
p53 in p53-deficient tumours can produce complete tumour
shRNA (W.X., L.Z. and S.W.L., unpublished data).
regressions. The primary response to p53 was not apoptosis, but
To determine whether transient p53 reactivation could also cause
instead involved the induction of a cellular senescence program
tumour regression, we treated transformed cells in culture or
that was associated with differentiation and the upregulation of
tumour-bearing mice with Dox for 4 days and then removed the
inflammatory cytokines. This program, although producing only
drug. Immunoblotting revealed that p53 could be transiently
cell cycle arrest in vitro, also triggered an innate immune response
induced following Dox addition and withdrawal (Fig. 1d). In cul-
that targeted the tumour cells in vivo, thereby contributing to
tured cells, even two days of Dox treatment reduced colony forma-
tumour clearance. Our study indicates that p53 loss can be
tion to levels observed following continuous Dox treatment
required for the maintenance of aggressive carcinomas, and illus-
(Supplementary Fig. 2a). Furthermore, both in situ and subcutan-
trates how the cellular senescence program can act together with
eous liver carcinomas showed complete regressions after only four
the innate immune system to potently limit tumour growth.
days of Dox treatment (Fig. 1e; Supplementary Fig. 2b). Thus, p53
p53 mutations are common in human liver cancer6, which is typ-
can induce tumour involution through a process that, once activated,
ically highly aggressive and resistant to non-surgical therapies. To
seems irreversible. These observations are analogous to results seen
determine the requirement for p53 loss in the maintenance of such
in murine tumours conditionally expressing various oncogenes,
carcinomas, we used reversible RNAi7 to control p53 in a chimaeric
where silencing of the initiating oncogene often causes tumour
liver cancer mouse model (Fig. 1a)4,5. Purified embryonic liver pro-
genitor cells (hepatoblasts) were transduced with retroviruses expres-
The rapid involution of hepatocarcinomas re-expressing p53 is
sing oncogenic ras (HrasV12), the tetracycline transactivator protein
consistent with p53's well-characterized ability to promote apopto-
tTA (‘tet-off') and a tet-responsive p53 miR30 design short hairpin
sis. We therefore examined apoptosis and proliferation in tumours
RNA (shRNA; Supplementary Fig. 1a)7,8, and seeded into the livers of
before and after p53 restoration (Fig. 2a, b). Surprisingly, we
athymic nude mice following intrasplenic injection4,5. To facilitate in
observed few cells that were TUNEL-positive or contained activated
vivo imaging, the oncogenic ras retrovirus co-expressed green fluor-
caspase 3 following p53 reactivation, suggesting that the primary
escent protein (GFP) and, in some experiments, hepatoblasts were
response to p53 was not apoptosis. Similarly, substantial necrosis
also co-transduced with a luciferase reporter.
was not observed in the regressing tumours. Instead, these tumours
p53 expression was efficiently suppressed in the absence of
showed a marked decrease in proliferation (Ki67) that was associated
doxycycline (Dox) and rapidly restored following Dox addition
with signs of cellular differentiation (Supplementary Fig. 3).
(Supplementary Fig. 1b, c). On transplantation into the livers of
p53 can also promote cellular senescence, an apparently irrevers-
recipient mice, hepatoblast populations co-expressing Ras and the
ible form of cell cycle arrest that is a potent barrier to tumori-
conditional p53 shRNA rapidly produced invasive hepatocarcinomas
genesis11–14 and can be triggered by hyperactive Ras or PI3K
in the absence of Dox (Fig. 1b), whereas cells expressing each vector
signalling12,15. Interestingly, hepatocarcinomas expressing either
alone did not (data not shown). These tumours were GFP-positive
oncogenic ras or Akt showed clear signs of senescence following p53
and, if expressing luciferase, could be visualized externally by bio-
reactivation in vivo (Fig. 3a–c; data not shown for Akt), including the
luminescence imaging (Fig. 1b).
accumulation of senescence-associated-b-galactosidase (SA-b-Gal)
1Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA. 2Howard Hughes Medical Institute, Cold Spring Harbor, New York 11724, USA. 3Division of MolecularPathology, Memorial Sloan-Kettering Cancer Center, New York 10021, USA.
*These authors contributed equally to this work.
2007
Nature Publishing Group

NATURE Vol 445 8 February 2007
activity (Fig. 3a, b) and the senescence markers p16INK4a, DcR2 and
inflammatory response, followed by destruction of tumour cells and
p15INK4b (Fig. 3c)13,15. SA-b-gal activity was also observed in tumours
following brief Dox treatment (Fig. 3b), indicating that a pulse of p53
Senescent cells often acquire a gene expression signature that
activity was sufficient to trigger senescence in vivo.
includes the upregulation of inflammatory cytokines and other im-
That p53 activation induces both cellular senescence and tumour
mune modulators16,17. Accordingly, inflammatory cytokines known
involution is surprising given that senescence is a cytostatic program.
to attract macrophages (Csf1 and Mcp1), neutrophils (Cxcl1) and
Indeed, transformed cells accumulated SA-b-gal activity but sub-
natural killer cells (Il15) were upregulated in liver tumours shortly
sequently remained arrested following p53 reactivation in vitro
following p53 reactivation (Fig. 4g). These genes were also induced
(Fig. 3d–f), suggesting that tumour regression involves non-cell-
by p53 in cultured hepatoma cells, demonstrating that they are
autonomous processes. Microscopic examination of tumours har-
expressed in tumour cells, not merely the infiltrating leukocytes
vested at different times following p53 reactivation revealed a
(Fig. 4g). Moreover, several adhesion molecules including Icam1
progressive inflammatory reaction involving polymorphonuclear
(ref. 18) and Vcam1 were induced following p53 reactivation
leukocytes, initially developing in peri-tumoral regions and ulti-
(Fig. 4g, and data not shown), indicating one way in which senescent
mately spreading throughout the tumour (Fig. 4a–f). We also
cells could facilitate immune recognition. Finally, transcripts
observed an intense perivascular infiltration in regressing tumours,
specific for neutrophils (Ncf2, Ncf4), macrophages (Mgl2, MSR2,
leading eventually to an overt vasculitis characterized by sclerosed
CD68) and natural killer cells (Klrb1, Klrd1) were increased in sen-
vessels, hemorraghia and erythrophagocytosis (Supplementary Fig.
escent tumours but not cultured cells. Thus, multiple components
7). Morphological, immunofluorescence and flow cytometric ana-
of the innate immune system infiltrate the tumours following p53
lyses identified the infiltrating leukocytes as neutrophils, macro-
phages and natural killer cells (Supplementary Figures 4–6). These
To determine whether innate immune cells were required for
histopathological features support a model of sequential events,
tumour clearance, mice bearing subcutaneous hepatocarcinomas
initiated by p53 reactivation in the tumour, activation of a dramatic
harbouring the conditional p53 shRNA were treated with gadolinium
Colour barMin = 1 × 105
Colour barMin = 1 × 105
Figure 1 Reactivation of p53 results in liver tumour regression.
progenitor cells with tet-off shRNA (TRE.shp53) or a non-regulatable
a, Embryonic liver progenitor cells were transduced with a tetracycline-
shRNA (MLS.shp53) were grown in nude mice. Values represent
regulatable p53 shRNA (TRE.shp53), tTA and H-rasV12. After onset of liver
mean 6 s.d. (n 5 4). d, p53 reactivation is reversed by Dox withdrawal.
tumours, p53 expression could be restored by doxycycline (Dox) treatment.
Protein lysates from liver progenitor cells pulse-treated with Dox for 4 days
b, Reactivation of p53 leads to rapid tumour regression. Tumour-bearing
were immunoblotted for p53. e, Representative mice (n 5 6) as in b were
mice were treated with Dox starting at day 0 and imaged at the indicated time
pulse-treated with Dox for 4 days and imaged at the indicated time.
points (n 5 9). c, Subcutaneous tumours derived from ras-transformed liver
2007 Nature Publishing Group


NATURE Vol 445 8 February 2007
chloride (a macrophage toxin)19,20 or neutralizing antibodies to sup-
reactivation in murine sarcomas also can induce senescence and
press neutrophil or natural killer cell function20,21, and monitored for
tumour clearance in a completely immunocompetent setting24.
tumour regression following Dox treatment. All three treatments
Our study used regulatable RNAi to demonstrate that p53 loss is
significantly delayed tumour regression following p53 reactivation
required for maintenance of aggressive hepatocarcinomas. We sus-
(Fig. 4i), thus confirming that components of the innate immune
pect that tumours harbouring p53 mutations may be hypersensitive
system were actively involved in tumour clearance, presumably
to restoration of p53 signalling because they have oncogenic lesions
through a coordinated response. Importantly, each antagonist effi-
or damage signals capable of potently activating p53 (refs 3,25). Still,
ciently and specifically depleted the targeted immune cells from the
the consequences of restoring p53 signalling may depend on tumour
spleen or peripheral blood (Supplementary Fig. 8), but did not pre-
origin or genotype; thus, whereas p53 reactivation induces sen-
vent tumour cell senescence (Fig. 4j). Similarly, almost no tumour
escence in liver carcinomas and sarcomas, lymphoid tumours
regression was observed following p53 reactivation in tumours
respond to p53 by undergoing apoptosis24. Tumours may also even-
grown in NOD/SCID mice —which have a highly impaired innate
tually escape their dependence on p53 mutations, but the fact that
immune system22—even though p53 still induced cytostasis and sen-
brief reactivation can cause complete tumour regressions supports
escence (Supplementary Fig. 9). Therefore, the delay in tumour clear-
the potential of transient p53 reactivation therapies26,27, even for
ance cannot be explained by a failure of the immune system to
advanced cancers.
phagocytose dead or dying cells. Instead, these results indicate that
Our results also identify a novel mechanism of tumour suppres-
the induction of cellular senescence and the evoked immune attack
sion involving cooperative interactions between a tumour cell sen-
cooperate to promote tumour clearance. Of note, the athymic nude
escence program and the innate immune system. They further
mice used here lacked functional B and T cells, which typically poten-
demonstrate that, despite the cytostatic nature of the senescence
tiate, but can also attenuate, inflammatory responses23. However, p53
program, senescent cells can turn over in vivo. Whether such turn-over is a general feature of senescence in vivo is not clear14,28, but whenpresent may reinforce the tumour suppressive action of senescence inpre-malignant settings or in tumours following treatment with sen-
escence- or differentiation-promoting therapies26,29. Conversely, ourresults identify a setting in which the innate immune system is pro-voked to coordinately attack tumour cells, presumably through both
phagocytosis and direct cytotoxic killing, thereby facilitating theirelimination. Although it is established that chronic inflammationtriggered by senescent stromal cells or other factors can promotetumorigenesis19,30, our study illustrates how innate immune cells—when targeted against senescent tumour cells—can have anti-tumoureffects as well. Strategies that specifically harness these processes may
represent a promising therapeutic approach.
β-Gal positive (%)
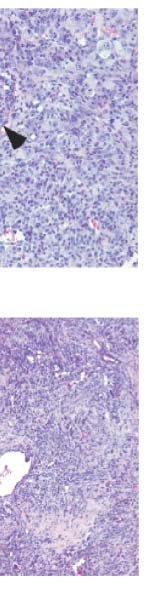
Figure 3 p53 reactivation induces cellular senescence. a, SA-b-Gal
staining of representative tumour-bearing livers untreated (p53 off) ortreated with Dox (p53 on, day 6) (n 5 3). b, SA-b-Gal staining of tumour
sections (n 5 3). Tumours were either untreated (p53 off), constantly
Figure 2 The primary response to p53 reactivation is not apoptosis.
treated with Dox for 8 days (p53 on 8 days) or briefly treated for 4 days and
a, Haematoxylin and eosin (H&E) immunohistochemical staining for
left untreated for 8 days (p53 on 4 days/off 8 days). Scale bar, 25 mm.
apoptotic cells (TUNEL and Caspase 3 staining) and proliferating cells (Ki67
c, Immunoblotting for senescence markers in normal liver or liver tumours
staining) of liver tumours before (p53 off) and after Dox treatment (p53 on).
treated with Dox for 0, 4 and 6 days. d, Liver progenitor cells harbouring ras
Tumours showed histopathology of human hepatocellular and
and tet-off shp53 were cultured in Dox-containing medium for 6 days (p53
cholangiocellular carcinoma. Inset (P) denotes positive controls (irradiated
on) and stained for SA-b-Gal. Values represent mean 6 s.d. (n 5 3;
thymus, 20Gy (20 Gray)). ‘p53 on' shows representative tumour on day 6.
**P , 0.0001). e, Representative pictures from d. f, Cells as in d were
Scale bar, 100 mm. b, Quantification of a. Values represent mean 6 s.d.
cultured with (p53 on, red line) or without Dox (p53 off, black line) and cell
(n 5 4; **P , 0.002).
numbers were counted. Values represent mean 6 s.d. (n 5 4).
2007 Nature Publishing Group



NATURE Vol 445 8 February 2007
tissue were fixed with 1% formalin for 1 min and stained for 12 h. Tumour-bearing livers were fixed with 4% formalin overnight and stained for 4 h.
Cultured cells were fixed with 4% formalin for 5 min and stained for 10 h.
RNA expression analyses. RNA isolation (Qiagen) and TaqMan reverse tran-
scriptase reaction (Applied Bosystems) were according to the manufacturer's
instructions. Quantitative PCR (qPCR) reactions (Bio-Rad) for each sample
were done in triplicate. Microarray experiments were performed on MouseGenome 430A 2.0 arrays (Affymetrix).
Data analysis. All the statistical analysis was done by Student's t-test.
Received 26 September; accepted 13 December 2006.
Published online 24 January 2007.
Harris, S. L. & Levine, A. J. The p53 pathway: positive and negative feedback loops.
Oncogene 24, 2899–2908 (2005).
Sherr, C. J. Principles of tumor suppression. Cell 116, 235–246 (2004).
Lowe, S. W., Cepero, E. & Evan, G. Intrinsic tumour suppression. Nature 432,
307–315 (2004).
Zender, L. et al. Generation and analysis of genetically defined liver carcinomas
derived from bipotential liver progenitors. Cold Spring Harb. Symp. Quant. Biol. 70,
251–261 (2005).
Zender, L. et al. Identification and validation of oncogenes in liver cancer using an
integrative oncogenomic approach. Cell 125, 1253–1267 (2006).
Below detection threshold
Staib, F., Hussain, S. P., Hofseth, L. J., Wang, X. W. & Harris, C. C. TP53 and liver
carcinogenesis. Hum. Mutat. 21, 201–216 (2003).
Dickins, R. A. et al. Probing tumor phenotypes using stable and regulated synthetic
microRNA precursors. Nature Genet. 37, 1289–1295 (2005).
Silva, J. M. et al. Second-generation shRNA libraries covering the mouse andhuman genomes. Nature Genet. 37, 1281–1288 (2005).
Immune cell transcript
Chin, L. et al. Essential role for oncogenic Ras in tumour maintenance. Nature 400,
468–472 (1999).
10. Jain, M. et al. Sustained loss of a neoplastic phenotype by brief inactivation of
MYC. Science 297, 102–104 (2002).
Braig, M. et al. Oncogene-induced senescence as an initial barrier in lymphoma
development. Nature 436, 660–665 (2005).
12. Chen, Z. et al. Crucial role of p53-dependent cellular senescence in suppression of
Isotype control Anti-NK
Pten-deficient tumorigenesis. Nature 436, 725–730 (2005).
13. Collado, M. et al. Tumour biology: senescence in premalignant tumours. Nature
436, 642 (2005).
Relative tumour volume
14. Michaloglou, C. et al. BRAFE600-associated senescence-like cell cycle arrest of
human naevi. Nature 436, 720–724 (2005).
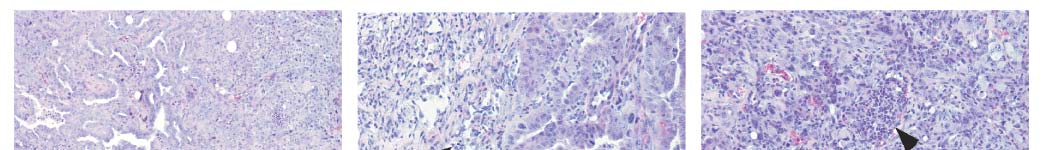
Figure 4 Clearance of liver tumours by an innate immune response.
15. Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras
a–f, Progressive immune infiltration in regressing tumours following p53
provokes premature cell senescence associated with accumulation of p53 andp16INK4a. Cell 88, 593–602 (1997).
reactivation (H&E; n 5 4). Arrows denote peri-tumoral polymorphonuclear
16. Minamino, T. et al. Ras induces vascular smooth muscle cell senescence and
(PMN) leukocytes. Arrowheads denote intra-tumoral polymorphonuclear
inflammation in human atherosclerosis. Circulation 108, 2264–2269
leukocytes. Scale bar, 100 mm. e, High magnification view of d. g, p53
reactivation leads to increased expression of chemokines and adhesion
17. Shelton, D. N., Chang, E., Whittier, P. S., Choi, D. & Funk, W. D. Microarray analysis
molecules in senescent tumours (in vivo) and cultured liver progenitor cells
of replicative senescence. Curr. Biol. 9, 939–945 (1999).
(in vitro). Values represent mean 6 s.d. (duplicate samples with triplicate
18. Gorgoulis, V. G. et al. p53-dependent ICAM-1 overexpression in senescent
qPCR). All the D4 and D8 values are statistically significant compared with
human cells identified in atherosclerotic lesions. Lab. Invest. 85, 502–511
D0 (P , 0.05). h, p53 reactivation is accompanied by increased immune cell
transcripts in senescent tumours (in vivo) but not in cultured cells (in vitro).
19. Maeda, S., Kamata, H., Luo, J. L., Leffert, H. & Karin, M. IKKb couples hepatocyte
Values represent the average of duplicate samples from microarrays.
death to cytokine-driven compensatory proliferation that promotes chemical
hepatocarcinogenesis. Cell 121, 977–990 (2005).
, Subcutaneous hepatocarcinomas were treated with Dox to induce tumour
20. Mundt, B. et al. Involvement of TRAIL and its receptors in viral hepatitis. FASEB J.
regression. The macrophage toxin GdCl (red line), an anti-neutrophil
17, 94–96 (2003).
antibody (purple) or an anti-natural killer-cell antibody (blue) was
21. Hong, F. et al. b-glucan functions as an adjuvant for monoclonal antibody
administered. Saline (solid black line) or an isotype control antibody
immunotherapy by recruiting tumoricidal granulocytes as killer cells. Cancer Res.
(dashed black line) served as controls. Values represent mean 6 s.d. (n 5 4;
63, 9023–9031 (2003).
*P , 0.02; **P , 0.002 for day 16). j, Blockade of innate immune cells does
22. Shultz, L. D. et al. Multiple defects in innate and adaptive immunologic function in
not prevent p53-induced senescence. Frozen sections from control animals
NOD/LtSz-scid mice. J. Immunol. 154, 180–191 (1995).
or immune antagonist treated animals were stained for SA-b-Gal activity
23. Ghiringhelli, F., Menard, C., Martin, F. & Zitvogel, L. The role of regulatory T cells in
(n 5 3). Scale bar, 50 mm.
the control of natural killer cells: relevance during tumor progression. Immunol.
Rev. 214, 229–238 (2006).
24. Ventura, A. et al. Restoration of p53 function leads to tumour regression
in vivo. Nature advance online publication, doi:10.1038/nature05541 (24 January2007).
See Supplementary Methods for detailed experimental methods.
25. Christophorou, M. A., Ringshausen, I., Finch, A. J., Swigart, L. B. & Evan, G. I. The
Generation and analysis of liver tumours. Isolation, culture and retroviral
pathological response to DNA damage does not contribute to p53-mediated
infection of murine hepatoblasts were as described4,5. Doxycycline treatment
tumour suppression. Nature 443, 214–217 (2006).
(BD Biosciences) and bioluminescence imaging (Xenogen) was performed
26. Bykov, V. J. et al. Restoration of the tumor suppressor function to mutant p53
according to the manufacturer's instructions. Histopathological evaluation
by a low-molecular-weight compound. Nature Med. 8, 282–288
was performed by an experienced pathologist (C.C.C.). Immunohisto-
27. Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule
chemistry was performed as described5.
antagonists of MDM2. Science 303, 844–848 (2004).
Tumour characterization. Fresh tumour tissue was lysed in Laemmli buffer
28. Gray-Schopfer, V. C. et al. Cellular senescence in naevi and immortalisation in
using a tissue homogenizer and analysed by immunoblotting by standard pro-
melanoma: a role for p16? Br. J. Cancer 95, 496–505 (2006).
cedures with the indicated antibodies. Detection of SA-b-gal activity was per-
29. Roninson, I. B. Tumor cell senescence in cancer treatment. Cancer Res. 63,
formed as described at pH 5.5 (ref. 7). Sections (10 mm) of snap frozen tumour
2705–2715 (2003).
2007 Nature Publishing Group
NATURE Vol 445 8 February 2007
30. Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent
Edith Seligson, the Don Monti Foundation, and grants from the National Institutes
fibroblasts promote epithelial cell growth and tumorigenesis: a link between
of Health (C.C.C, S.W.L.). This work is dedicated to our friend and colleague Dr.
cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077
Enrique (Henry) Cepero.
Author Contributions W.X.: study design and conduction of experiments; L.Z.:
Supplementary Information is linked to the online version of the paper at
study design and conduction of experiments; C.M.: design and conduction of flow
cytometry experiments; R.A.D.: vector development; E.H.: histopathologicalanalyses; V.K.: microarray analysis; C.C.C: histopathological analyses; S.W.L.:
Acknowledgements We thank L. Bianco and M. Jiao for technical assistance. We
study design, principal investigator.
also thank G. Evan, T. Jacks, A. Ventura, M. Narita, A. Chicas, M. Yon, G. Hannonand other members of the Lowe and Hannon laboratories for advice and
Author Information Reprints and permissions information is available at
discussions. We thank M. McCurrach for editorial assistance. W.X. is in the MCB
www.nature.com/reprints. The authors declare no competing financial interests.
graduate program at Stony Brook University. This work was generously supported
Correspondence and requests for materials should be addressed to S.W.L.
by the Emmy Noether Programme of the German Research Foundation, Alan and
2007 Nature Publishing Group
Source: http://blog.tcu.edu.tw/gallery/9/Senescence%20and%20tumour%20clearance%20is%20triggered%20by%20p53.pdf
15 april 2010 #16 www.erasmusmagazine.nl OP DE ARBEIDSMARKT Ontbijten met Wat nou 24/7 10 ‘Hypotheekrenteaftrek is pervers' Begin deze maand gaf ik een nieuwe collega een rondlei- ding op campus Woudestein. Hij komt uit Leiden, dus dan duik je als Rotterdammer toch wat in de excuusmodus als je zo langs onze grauwe gebouwen loopt. Weinig
Cellular and molecular effects of steroid hormones on CNS excitability SHERYL S. SMITH, PHD, AND CATHERINE S. WOOLLEY, PHD ■ ABSTRACT has been shown to have activating effects on mood The steroid hormones 17β-estradiol (estradiol) and (euphoria, anxiety, or antidepressant effects), cogni-tion, sensory response, motor behavior, and seizure



