Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Astra3 23.3
Translating cell biology into
therapeutic advances
in Alzheimer's disease
Dennis J. Selkoe
Studies of the molecular basis of Alzheimer's disease exemplify the increasingly blurred distinction between basic and
applied biomedical research. The four genes so far implicated in familial Alzheimer's disease have each been shown to
elevate brain levels of the self-aggregating amyloid-b protein, leading gradually to profound neuronal and glial
alteration, synaptic loss and dementia. Progress in understanding this cascade has helped to identify specific
therapeutic targets and provides a model for elucidating other neurodegenerative disorders.
Until the last decade, degenerative diseases of the brain were
are related to the pathogenesis of one or both of the classical lesions.
considered to be among the most obscure and intractable disorders
Early attempts to purify the tangle and plaque proteins and
in medicine. Alzheimer's disease (AD) epitomized the mechanistic
determine their respective compositions were met with consider-
ignorance and therapeutic nihilism that pervaded the study of
able scepticism as it was argued that, because the plaques and tangles
neurodegeneration in humans. But research advances in two
were end-stage lesions that apparently represented the tombstones
broad areas—biochemical pathology and molecular genetics—
of the pathogenic process, such knowledge would provide little
have combined to offer new hope and to stimulate research.
useful information about aetiology and early pathogenesis. It has
Determining the composition of the classical brain lesions and
become apparent in recent years that this concern was ill-founded.
identifying at least four genes that predispose individuals to the
Neuritic plaques are spherical, multicellular lesions that are
disorder have increased our understanding of the genotype-to-
usually found in moderate or large numbers in limbic structures
phenotype relationships that underlie inherited forms of AD. It is
and association neocortex (reviewed in ref. 1). They contain extra-
therefore timely to review and attempt to integrate the disparate
cellular deposits of amyloid-b protein (Ab) that include abundant
elements of the disease into a coherent whole, perhaps helping to
amyloid fibrils (7–10 nm) intermixed with non-fibrillar forms of
focus future investigative efforts on developing rational treatments.
this peptide. Considered to be mature lesions that are generallyassociated with full-blown clinical disease, neuritic plaques have
Three central questions about Alzheimer's syndrome
degenerating axons and dendrites (neurites) within and intimately
It is now clear that AD is, in reality, a multifactorial syndrome,
surrounding the amyloid deposit (Fig. 1). Such plaques also contain
rather than a single disease. Given the complex array of factors that
variable numbers of activated microglia that are often situated
may initiate or propagate the syndrome, it sometimes seems that
within and near the fibrillar amyloid core, as well as reactive
researchers are pursuing many ostensibly unrelated clues to itspathogenesis. Yet almost all investigations of AD during the pasttwo decades have sought to provide information about one or moreof three interrelated questions. First, what are the causes of thedisorder? Second, regardless of cause, is there a common cellbiological and biochemical mechanism that leads to the dementiain essentially all cases? And third, what is the phenotype of thedegenerating neurons affected by this mechanism: where are theylocated, what are their neural connections and neurotransmitterspecificities, and what behavioural symptoms do they mediate?When thought of in this way, the profusion of initially distinctobservations about the syndrome can be evaluated with respect tothe particular step in the disease cascade that each study addresses.
Here I review the progress made in attempting to synthesize theanswers to these questions into a mechanistic pathway. In doing so, Ihope to show that the elucidation of AD represents an emergingtriumph of reductionist biology applied to a chronic disorder of themost complex of biological systems, the human cerebral cortex.
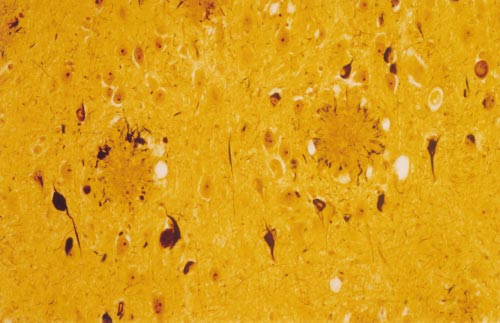
Figure 1 High-power photomicrograph of a section of the amygdala from an
Biochemistry of the classical brain lesions
Alzheimer's patient showing the classical neuropathological lesions of the
Senile (neuritic) plaques and neurofibrillary tangles, observed in
disorder. The modified Bielschowsky silver stain demonstrates two senile
Alzheimer's original patient of 1906, comprise the major neuro-
(neuritic) plaques consisting of compacted, spherical deposits of extracellular
pathological lesions that, when present in sufficient numbers in
amyloid immediately surrounded by a halo of silver-positive dystrophic neurites,
limbic and association cortices, allow a definitive diagnosis of AD
which can include both axonal terminals and dendrites. Some of the pyramidal
after the patient's death (Fig. 1). Although there are other distinct
neurons in this field contain neurofibrillary tangles, which are darkly staining
pathological changes that often appear to be physically separate
masses of abnormal filaments occupying much of the perinuclear cytoplasm.
from the plaques and tangles (for example, microvascular amyloidosis
Electron microscopy of such neurons generally reveals large, non-membrane-
and dystrophic cortical neurites), evidence indicates that even these
bound bundles of paired helical filaments.
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
astrocytes surrounding the core. Immunohistochemistry using
dominant pattern. Such factors can therefore be difficult to recognize
antibodies against Ab reveals an even larger number of deposits
in epidemiological studies.
in the Alzheimer's brain that seem to lack altered microglia and
At present, there are four well confirmed genes in which muta-
astrocytes and surrounding dystrophic neurites. These lesions are
tions or polymorphisms can result in AD, and several other
referred to as diffuse plaques, and within these the Ab occurs in a
candidates are in various stages of confirmation. The first AD-
predominantly non-fibrillar, amorphous form in the neuropil2.
causing gene to be identified was that encoding the precursor of Ab,
Diffuse deposits are almost exclusively composed of the highly
the b-amyloid precursor protein (APP). Missense mutations in APP
amyloidogenic 42-amino-acid-residue form of the peptide (Ab42).
account for a tiny fraction (less than 0.1%) of all Alzheimer's cases,
This form is normally produced by cells in much lower quantities
but they have proved to be highly informative as regards the
than the 40-residue form (Ab40), which represents roughly 90% of
pathogenic mechanisms of AD in general. For example, expression
total secreted Ab. Ab deposits do not occur simply in these two
of mutant APP transgenically in mice provided the first repro-
extreme forms (diffuse and neuritic), but rather as a continuum in
ducible and robust animal models of the disease. Inheritance of one
which mixtures of fibrillar and non-filamentous forms of the
or two e4 alleles of ApoE is a far more prevalent genetic basis for AD.
peptide can be associated with varying degrees of local glial and
ApoE4 helps precipitate the disorder primarily in subjects in their
sixties and seventies, thus lowering the typical age of late-onset AD5.
In regions of the Alzheimer brain that are generally not impli-
There is also evidence that an alternative ApoE allele, e2, confers
cated in the clinical syndrome, for example the cerebellum and
some protection from the development of AD. It should be
thalamus, almost all Ab deposits seem to be diffuse, with little
emphasized that ApoE4 is a risk factor for, not an invariant cause
evidence of local glial and neuritic reaction. Likewise, the brains of
of, AD. Some humans who are homozygous for this isoform
aged, cognitively normal humans often contain Ab deposits, but
continue to show no Alzheimer symptoms in their nineties. The
these are primarily of the diffuse type, with few neuritic plaques and
third and fourth genes implicated in familial forms of AD are
neurofibrillary tangles present in limbic and association cortices.
designated presenilin-1 (PS1) and presenilin-2 (PS2), because
Ab also accumulates in the basement membranes of some cerebral
missense mutations result in an aggressive, early-onset form of
capillaries, arterioles and venules and some meningeal arterioles.
the disorder, usually beginning between the age of 40 and 60
The extent of this microvascular b-amyloidosis usually does not
years6–8. PS1 and PS2 are homologous polytopic proteins that are
correlate closely with the number of Ab plaques in a brain, and its
believed to span certain membranes of cells eight times (see below).
importance in contributing to the dementia remains a subject of
More than 50 missense mutations have been identified in PS1 and
active research.
2–3 in PS2; these are widely scattered in the molecule but tend to
Neurofibrillary tangles are intraneuronal cytoplasmic lesions
cluster within and adjacent to the transmembrane domains.
consisting of non-membrane-bound bundles of paired, helically
The inheritance of a polymorphism in the gene encoding a2-
wound ,10-nm filaments (PHF), sometimes interspersed with
macroglobulin, a large multifunctional protein that can act as a
straight filaments3. Neurofibrillary tangles generally occur in large
special kind of protease inhibitor, has been associated with
numbers in the Alzheimer brain, particularly in entorhinal cortex,
increased risk of late-onset AD9, and genetic epidemiological studies
hippocampus, amygdala, association cortices of the frontal, tem-
are underway to confirm the occurrence and frequency of this
poral and parietal lobes, and certain subcortical nuclei that project
polymorphism in AD. Families with multiple AD members that
to these regions. The subunit protein of the PHF is the microtubule-
show no linkage to any of the above five genes are also under study
associated protein, tau. Biochemical studies have shown that the tau
in an attempt to identify or confirm additional genetic risk factors
found in the tangles and also in many of the dystrophic neurites
or autosomal dominant mutations. Within a decade or two, a
within and outside the plaques comprises hyperphosphorylated,
sizeable number of additional genes will be implicated, most of
insoluble forms of this normally highly soluble cytosolic protein.
them probably acting as polymorphic risk factors in some populations.
The insoluble tau aggregates in the tangles are often conjugated with
Despite the prominence of tau accumulation in the neurofibrillary
ubiquitin, a feature they share with other intraneuronal protein-
tangles and dystrophic neurites of a high percentage of AD cases, the
aceous inclusions in aetiologically diverse disorders such as Parkin-
tau gene has so far not been found to be the site of mutations in
son's disease and diffuse Lewy-body disease. If this ubiquitination
familial AD. Instead, mutations in tau have been discovered in
represents an attempt to remove the tau filaments by way of the
families with a less common dementia: frontotemporal dementia
proteasome, it seems to be largely unsuccessful. Tangles also occur
with parkinsonism linked to chromosome 17 (FTDP-17)10–12. This
in more than a dozen relatively uncommon neurodegenerative
disorder is characterized by widespread neurofibrillary tangle for-
diseases in which one usually finds no Ab deposits and neuritic
mation associated with specific biochemical alterations in the
microtubule-binding properties of tau13 in the absence of amyloid
Therefore, the two classical lesions of AD can occur indepen-
deposits. The discovery of tau mutations in this distinct form of
dently of each other. As I shall discuss, there is growing evidence that
dementia proves that a primary alteration of tau structure and
the formation of tangles represents one of several cytological
function can lead to progressive, severe neuronal degeneration and,
responses by cells to the gradual accumulation of Ab and Ab-
ultimately, to the death of the patient. This finding also shows that
even severe neurofibrillary tangle formation does not lead tosecondary accumulation of Ab as diffuse and neuritic plaques.
The genetics of Alzheimer's disease
The latter point addresses a recurring controversy in the study of
It has been known for several decades that AD can occur in a familial
AD, that is, whether plaques or tangles have temporal precedence in
form that transmits as an autosomal dominant trait. Estimates of
the pathogenesis of the disorder. Both the APP and presenilin
the proportion of Alzheimer's cases that are genetically based have
mutations in AD and the tau mutations in FTDP-17 support the
varied widely from as low as 10% to as high as 40 or 50%, and some
conclusion that the tau alteration in AD follows Ab accumulation
investigators believe that almost all cases will be shown eventually to
rather than vice versa.
have genetic determinants. It is difficult to resolve this question in alate-onset disorder that, up until the past two decades, was often not
Cell biology of APP
explicitly diagnosed. Moreover, the discovery that the e4 allele of
During the past few years, progress in understanding the transport
apolipoprotein E (ApoE) is a normal polymorphism that confers
and unusual proteolytic processing of APP, together with the
increased risk for developing AD4 indicates that genetic factors
importance of the presenilins in these pathways, has provided
predisposing individuals to AD need not occur in a simple, autosomal
insights into the molecular basis of familial AD. The results of
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
Figure 2 Diagrams of APP and its principal metabolic derivatives. The upper
proteolytic cleavage made by a protease(s) designated a-secretase, which
diagram depicts the largest of the known APP alternate splice forms, comprising
enables secretion of the large, soluble ectodomain (sAPP-a) into the medium and
770 amino acids. Regions of interest are indicated at their correct relative
retention of the 83-residue C-terminal fragment (C83) in the membrane. The latter
positions in the linear sequence. A 17-residue signal peptide occurs at the N
can undergo cleavage by the activity termed g-secretase at residues 711 or 713 to
terminus. An alternatively spliced exon of 56 amino acids is inserted at residue
release the p340 and p342 peptides. The lower diagram depicts the alternative
289; it contains a serine-protease-inhibitor domain of the Kunitz type (KPI). A
proteolytic cleavage after residue 671 by the activity termed b-secretase. This
single membrane-spanning domain (TM) at amino acids 700–723 is indicated by
results in the secretion of the slightly truncated sAPP-b molecule and the
the vertical dashed lines. The Ab region is indicated in red. In the middle diagram,
retention of C99. The latter can also undergo cleavage by g-secretase at 711 or 713
the left arrow indicates the site (after residue 687; this is the same site as that
to release the Ab40 and Ab42 peptides.
indicated by the white dot in the Ab region of the upper diagram) of a constitutive
these studies have implications also for the mechanism of the
isoforms), enhancement of cell-substrate adhesion, neuritotrophic
phenotypically similar ‘sporadic' form of the disease and have
provided new information about fundamental features of protein
properties16. No evidence has emerged that a fundamental cellular
structure and function.
function of APP is lost in AD patients; instead, APP mutations seem
The cloning of the gene on chromosome 21 that encodes APP14
to act by a gain-of-function mechanism, namely the increased
was made possible by the purification and sequencing of its Ab
production of the potentially cytotoxic Ab fragment (see below).
fragment from the microvascular amyloid deposits of AD and
APP has one ,23-residue hydrophobic stretch near its carboxy-
Down's syndrome patients15. APP comprises a group of ubiquitously
terminal region (Fig. 2) that anchors it in internal membranes (for
expressed polypeptides whose heterogeneity arises from both alter-
example, endoplasmic reticulum (ER), Golgi, trans-Golgi network
native splicing and post-translational processing (reviewed in
and endosome) and in the plasmalemma. Both during and after its
ref. 16). In addition to the 751- and 770-residue splice forms
transport through the secretory pathway to the cell surface, a subset
expressed in non-neuronal cells throughout the body, neurons
of APP molecules undergoes specific endoproteolytic cleavages,
express a more abundant 695-residue isoform. The difference
most frequently by a scission between amino acids 16 and 17 of
between the 751/770- and the 695-residue forms is the presence in
the Ab region, that is, 12 residues amino terminal to the transmem-
the former of an exon that codes for a 56-amino-acid motif that is
brane sequence (Fig. 2). This principal secretory cleavage is effected
homologous to the Kunitz-type of serine protease inhibitors,
by a protease(s) designated a-secretase(s). The cut creates a large,
indicating one potential function of these longer APP isoforms.
soluble ectodomain fragment (sAPP-a) that is released into vesicle
Indeed, the 751/770 forms of APP present in human platelets serve
lumens and from the cell surface, and a membrane-retained
as inhibitors of Factor XIa (a serine protease) in the coagulation
C-terminal fragment (CTF) of 83 amino acids (C83) (Fig. 2).
cascade. Nevertheless, deletion of the gene in mice results in neither
a-Secretase(s) are probably membrane-anchored proteases capable
early mortality nor appreciable morbidity; cerebral gliosis and
of cleaving diverse single transmembrane proteins, and they seem to
changes in locomotor behaviour occur later in adult life17, and
cleave APP at a specific distance from the outer membrane surface
neurons cultured at birth may have diminished viability and
while showing little sequence specificity21. Although the constitutive
retarded neurite outgrowth18.
a-secretases are not yet clearly defined, the regulated cleavage of
The lack of a vital consequence of APP deletion in vivo may result
APP (for example, as enhanced by phorbol esters) may be carried
from mammals expressing proteins that are closely homologous to
out by certain metalloprotease disintegrins that are capable of
APP—the amyloid precursor-like proteins (APLPs)19,20—but
shedding the ectodomains of proteins such as tumour-necrosis
which do not contain the Ab sequence. Although some activities
factor-a (TNF-a)22,23. In most cell types, a minority of all APP
of holoAPP or its major secreted derivative, sAPP-a, have been
molecules undergoes a-secretory cleavage, so that any increase in
inferred using cell-culture studies, the principal function(s) of the
this scission would still leave many APP polypeptides that could be
molecule in vivo remain unclear. Functions that have been described
subjected to the alternative cleavages (made by the b- and
in vitro include inhibition of certain serine proteases (for the APP751/770
g-secretases) that lead to Ab formation.
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
The generation of amyloid-b protein
Cell biology of the presenilins
Ab is secreted constitutively by normal cells in culture and detected
PS1 and PS2 are homologous, polytopic membrane proteins that
as a circulating peptide in the plasma and cerebrospinal fluid (CSF)
have been localized so far to ER and Golgi in mammals. A member
of healthy humans and other mammals24–27. When this unexpected
of the Caenorhabditis elegans presenilin family, sel-12, is a facilitator
observation was made in 1992, it was also recognized that a smaller
of lin-12/Notch signalling during the determination of cell fate in
fragment, which had a relative molecular mass of 3,000 (Mr 3K) and
development29. Wild-type human PS1 can rescue the lethal pheno-
which comprised the latter two-thirds of Ab (designated p3), was
type caused by sel-12 mutations in C. elegans, whereas most AD-
released constitutively by APP-expressing cells during normal
linked mutant PS1 molecules examined in this bioassay confer only
metabolism24. These and other studies have shown that the N
partial functional recovery30,31. Despite their structural and functional
terminus of p3 is generated when a-secretase cleaves APP, and its
homologies, the precise cellular activities of the presenilin/sel-12
C terminus is generated when the resultant C83 CTF is cleaved by
proteins are not yet known (but see later). PS1 can interact with a
the unusual activity referred to as g-secretase(s) (Fig. 2). In an
novel neuron-specific member of the Armadillo family, d-catenin,
analogous fashion, other APP holoproteins are instead cleaved
in the yeast two-hybrid system, and both d- and b-catenins co-
by b-secretase just before the Ab region to create its N terminus,
immunoprecipitate with PS1 (refs 32, 33). These results indicate
followed by cleavage of the resultant 99-residue CTF (C99) by
that, in addition to facilitating Notch activity, PS1 may interact with
g-secretase(s) to create Ab (Fig. 2). The scission by b-secretase members of the Armadillo family that are known to serve asreleases a truncated form of sAPP (sAPP-b) from the cell28.
intracellular components of cell–cell adhesion complexes. Deletion
Although precise quantification is not available, it seems that a
of the PS1 gene in mice produces an embryonic-lethal phenotype
substantially smaller portion of total cellular APP undergoes
characterized by severely disordered somitogenesis and axial skeletal
cleavage by b- than by a-secretase. Moreover, not all of the resultant
development34,35 as well as by neurodevelopmental changes in the
C99 and C83 fragments are processed by g-secretase to Ab and p3,
forebrain35. Both wild-type and AD-linked mutant PS1 can rescue
respectively; alternative proteolytic pathways can fully degrade these
this knockout phenotype in mice36. Thus, the AD-linked presenilin
CTFs, probably in late endosomes and lysosomes (Box 1).
mutations do not confer loss of function in mammals, but insteadresult in a dominantly transmitted gain of function.
Presenilins are expressed at low abundance in most cell types,
Box 1 Complexity of the amyloidogenic processing of APP
including neurons. Steady-state levels of the presenilin holoproteins
Ab peptides present in culture medium, human CSF and the brain amyloid
are low because the precursor undergoes endoproteolysis to
deposits of AD subjects show heterogeneity in both their amino- and
generate stable N- and C-terminal fragments37. The constitutive
carboxy-terminal regions24,26,88–90. The C-terminal heterogeneity of Ab has
proteolytic cleavage site38 occurs in a hydrophobic portion of the
special importance for its aggregation. Immunohistochemistry with anti-
cytoplasmic loop between the sixth and seventh of the eight39
bodies that selectively recognize either the Val 40 or the Ala 42 C terminus
transmembrane (TM) domains believed to exist in PS1. The
have revealed that the first Ab form deposited as diffuse plaques in AD
steady-state levels of the presenilin N-terminal fragments (NTFs)
and Down's syndrome brains ends at residue 42 (refs 47, 90). In studies of
and CTFs seem to be tightly regulated, as overexpression of PS1 in
the temporal progression of plaque formation in the brains of Down's
transfected cells or transgenic mice generally does not increase the
syndrome patients of increasing age, Ab42 peptides can form numerous
levels of the fragments40; the excess holoproteins are rapidly
diffuse plaques as early as age 12 years, whereas Ab40 is first detected in
degraded, mainly by the proteasome41. Once formed, the PS1
the plaques almost 20 years later47. This evidence of initial Ab42 deposition
fragments associate into higher molecular mass (,150K) com-
in AD and Down's syndrome brains fits well with biochemical studies91
plexes that may represent the principal form in which presenilin
showing that the Ab42 peptide, with its two additional hydrophobic
functions in cells33,42. Subcellular fractionation indicates that the
residues, aggregates far more rapidly into amyloid fibrils (as well as into
PS1 holoprotein is found principally in ER vesicles, where the
intermediate assemblies called protofibrils84,85) than does the Ab40 pep-
constitutive endoproteolysis can first be detected; the fragments
accumulate subsequently in Golgi-type vesicles, where they are
Because the b- and g-secretases have yet to be identified definitively, it
highly stable43.
is difficult to determine their precise location in the cell. A portion of Ab
By using the yeast two-hybrid system and/or co-immuno-
peptides appears to be generated in recycling endosomes after
precipitation, several known or newly identified proteins have
internalization of APP molecules from the cell surface. That surface
been shown to interact in vitro with PS1 or PS2, but their
APP can indeed undergo clathrin-mediated endocytosis and then recycle
importance for the normal and pathogenic functions of the pre-
rapidly to the surface has been established in both non-neural92 and
senilins is unclear. In particular, those proteins that interact with
neuronal93 cells. The clearest evidence that Ab can be generated from
either PS1 or PS2 alone seem unlikely to be crucial in the pathogenic
reinternalized APP molecules has come from experiments in which APP
mechanism of the presenilins in AD, because mutations of con-
on the plasma membrane of intact cells was radioiodinated and allowed
served residues in both proteins produce elevation of Ab42
to internalize at 37 8C; this led to the release within 15–30 minutes of
production44 and lead to a similar clinicopathological phenotype.
radioiodinated Ab that could have arisen only from the surface-labelled
Sequences that diverge between PS1 and PS2 (such as the
molecules94. As regards the loci for Ab42 formation, the chemical retarda-
distal TM6 → TM7 ‘loop' domain of PS1, which binds the catenins)
tion of APP transport through the secretory pathway and the use of
are less likely to be required for the critical stabilization of the
sensitive Ab enzyme-linked immunosorbent assays (ELISAs) on isolated
presenilin heterodimers and for their AD-promoting activity than
vesicle fractions have suggested that Ab42 can be generated early during
are highly conserved sequences (such as the C terminus45).
secretory processing (for example, in the ER and Golgi)62,95,96. Direct
quantification of both Ab peptides in subcellular fractions indicates that
Ab42 is the most abundant species in ER-rich fractions, whereas more
Cultured cells and transgenic mice have been used to model the
Ab40 than Ab42 is detectable in Golgi-rich fractions62. That Ab can be
biochemical and neuropathological effects of each of the four genes
generated and/or accumulate at various points during the secretory
implicated so far in familial AD. The results have been compared to
processing of APP is further supported by biochemical and immuno-
the actual phenotypes observed in the brains of patients with the
cytochemical experiments detecting sAPP-b in ER97 and post-Golgi
respective gene defects. In all four cases, inherited alterations in the
secretory vesicles98, Ab42 in ER99 and Ab peptides in detergent-insoluble
gene products have been linked to increases in the cerebral produc-
glycolipid (DIG) membranes100.
tion and/or deposition of the Ab peptides (reviewed in ref. 46). Thiswork has provided strong support for the importance of cerebral Ab
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
accumulation as an early, invariant and necessary event in the
tangle formation associated with progressive dementia and severe
genesis of familial AD.
Alterations in the APP gene can lead to the AD syndrome in at
haemorrhages50. These adjacent mutations within the Ab sequence
least two ways: either by overexpression (owing to a gene dosage
may alter a-secretase processing to favour b-secretase cleavage of
effect in trisomy 21 (Down's syndrome)) or by missense mutations
APP, but they are also likely to increase the propensity of the mutant
that increase the amyloidogenic cleavages of APP at either the b-
peptides to aggregate into amyloid fibrils. In short, the APP
secretase site (resulting in excessive production of both Ab40 and
mutations result in increased production and deposition of Ab in
Ab42) or the g-secretase site (resulting in selectively increased
the brain and its microvasculature. No other APP mutations away
production of Ab42). In trisomy 21, a lifelong increase in APP
from the sites of the secretase cleavages have been discovered in AD
expression and the resultant overproduction of both Ab40 and Ab42
families. If APP mutations caused familial AD by perturbing the
peptides is assumed to be responsible for the early appearance of
normal function of the precursor (as has sometimes been hypothe-
some or many Ab42 diffuse plaques, which occur as early as age 12
sized), then one would expect AD-linked mutations to be more
years and accumulate with time. Because Down's patients invariably
widely distributed in the molecule, not exclusively clustered at the
develop the full-blown neuropathology of AD by their forties or
fifties, the temporal progression of AD-type lesions between the
In the case of the ApoE4 polymorphism, co-expression of each of
early teens and the forties has been considered to represent the
the three human ApoE alleles with APP in cultured cells shows no
sequence of pathogenesis in conventional AD. Down's subjects often
differential change in the proteolytic processing of APP to Ab
display diffuse plaques composed solely of Ab42 in their teens and
(ref. 51). Instead, the disease-promoting effect of inheriting one
twenties, with accrual of Ab40 peptides onto these plaques and the
or two ApoE4 alleles seems to involve enhanced aggregation and/or
appearance of associated microgliosis, astrocytosis and surrounding
decreased clearance of Ab (refs 52–55). The resultant increase in
neuritic dystrophy beginning in their late twenties or thirties47,48.
steady-state levels of cerebral Ab has been demonstrated by crossing
This observation exemplifies the importance of Ab42 deposition as a
APP transgenic mice with knockout mice that lack ApoE: far less Ab
potentially seminal event in the development of AD pathology. The
plaque formation is observed in these offspring than when ApoE is
appearance of neurofibrillary tangles is also delayed until the late
present in the brain56.
twenties or thirties in most Down's patients. The gradual accrual of
Perhaps the most interesting genotype-to-phenotype relation-
AD-type brain lesions in these individuals (who are retarded from
ship in AD involves the presenilin mutations. Even before PS1 and
birth for other reasons) appears to be associated in many cases with
PS2 mutations were expressed in cultured cells and transgenic mice,
further loss of cognitive and behavioural functions after the age of
assays of Ab40 and Ab42 in the plasma and skin fibroblast media of
humans bearing these mutations revealed a selective 1.5–3-fold
All eight reported APP missense mutations linked to AD are
elevation in Ab42 (ref. 44). Modelling these mutations in vitro and in
clustered at the b-secretase cleavage site, just after the a-secretase
vivo confirmed this result (reviewed in ref. 57). Indeed, crossing
site or just after the g-secretase site (Fig. 3). Studies in cell cultures
mice transgenic for mutant APP with mice expressing a PS1
and transgenic mice have shown that these mutations enhance
mutation results in a substantially accelerated AD-like phenotype,
either the b-secretase or the g-secretase cleavage of APP, resulting in
with Ab42 plaques (both diffuse and mature) occurring as early as
chronically elevated levels of Ab42. For the two missense mutations
3–4 months of age58. Moreover, the ability of presenilin mutations
located internally in Ab (Fig. 3), one produces particularly severe
selectively to enhance Ab42 deposition in the brain has been
microvascular b-amyloidosis with relatively minor parenchymal
demonstrated directly in patients carrying these mutations59,60.
deposition (the E22Q mutation, in which glutamine is substituted
How do mutations in the eight-transmembrane (TM) presenilin
for glutamic acid at position 22 and which causes hereditary
proteins cause selective Ab42 hypersecretion? Two broad hypotheses
cerebral haemorrhage with amyloidosis of the Dutch type)49. The
about the mechanism have emerged. One suggests that the presenilin
other mutation (glycine substituted for alanine at position 21
molecule regulates the transport of g-secretase or APP to each other
(A21G)) leads to a mixed phenotype of AD-type plaque and
without any physical interaction with APP. This mechanism has been
Figure 3 APP mutations causing AD or hereditary cerebral haemorrhage. The
mutations linked to familial AD, and their respective codon numbers are given
wild-type sequence of Ab and regions that immediately flank it in human APP is
above the sequence. The major sites of the b-, a- and g-secretase cleavages are
shown by the single-letter code. The underlining indicates the Ab1–42 peptide. The
bold residues below the wild-type sequence indicate the reported missense
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
suggested by investigators who have failed to co-immunoprecipitate
The inflammatory and neurotoxic cascade
APP with PS1 (ref. 61). The alternate hypothesis, physical involve-
I have focused on some of the ways in which genetic alterations can
ment of the presenilins in the g-secretase cleavage of APP, has been
lead to chronically elevated concentrations of Ab in the brain, but
proposed by those who have detected small amounts of APP
the putative downstream effects of this accumulation remain the
co-precipitating with presenilin in whole-cell lysates and in isolated
subjects of intensive study (Fig. 4). Chronic elevation of Ab42 in
ER and Golgi vesicles62–64. If presenilins were required to bring APP
brain interstitial fluid (and perhaps also inside neurons69), caused
and g-secretase together without actually contacting APP, this
by defects in the genes encoding APP and the presenilins, is assumed
transport role must involve membranous microdomains within
to lead gradually to oligomerization and, eventually, fibrillization of
a subclass of vesicles, as it has already been shown that APP, PS1
the peptide and its deposition as diffuse and, later, mature plaques.
and the g-secretase-mediated products Ab
Based on studies of Down's syndrome patients and transgenic mice
42 and Ab40 can all be
recovered together within purified, calnexin-rich ER vesicles43,62.
that express mutant APP and/or PS1, it is hypothesized that Ab42
Evidence has emerged recently that supports a direct involvement
accumulation and diffuse plaque formation is associated with local
of presenilins in the g-secretase cleavage of APP65. Because mice
microglial activation, cytokine release, reactive astrocytosis and a
deficient in PS1 show markedly decreased g-secretase processing of
multi-protein inflammatory response70–72, including the binding of
C99 to Ab (ref. 66), it seemed possible that PS1 and PS2 could
the C1q component of the classical complement cascade by Ab
themselves be g-secretases. However, the multi-transmembrane
(ref. 73) and the triggering of this cascade74. It has been proposed
structure of the presenilins made them unlikely candidates for
that such a glial inflammatory process and/or any direct neurotoxic
proteases. Nevertheless, the recognition that all members of the
effects of oligomeric and fibrillar Ab could produce the multifaceted
presenilin gene family have two aspartic acid residues in TM6 and
biochemical and structural changes in surrounding axons, dendrites
TM7, respectively, which flank the constitutive presenilin cleavage
and neuronal cell bodies that characterize the limbic and association
site in the proximal part of the TM6 → TM7 cytoplasmic loop, led
cortices in AD. There is considerable evidence that the effects of an
to the hypothesis that these residues might represent the active site
Ab-initiated inflammatory and neurotoxic process include exces-
of an unprecedented intramembranous aspartyl protease65. Mutation
sive generation of free radicals and peroxidative injury to proteins
of either transmembrane aspartate to alanine resulted in both the
and other macromolecules in neurons75,76. In this regard, a thera-
abolition of presenilin endoproteolysis and the marked inhibition
peutic trial of the antioxidant, vitamin E, seemed to result in slower
of g-secretase processing of C99 to Ab (and C83 processing to p3)65.
clinical progression of the disease, although actual amnestic symptoms
Apparently, the Asp → Ala mutant isoforms, when transfected into
were not noticeably improved77. Among the many possible meta-
several cell types, act as dominant negatives to suppress endogenous
bolic consequences of Ab accumulation and aggregation, altered
PS1 and obviate both PS endoproteolysis and g-secretase cleavage of
ionic homeostasis, particularly excessive calcium entry into neurons,
C99 and C83. These unexpected results indicate that the two trans-
could well contribute to selective neuronal dysfunction and cell
membrane aspartates either allow presenilins to serve as essential
death, based on studies of the in vitro effects of aggregated Ab
diaspartyl co-factors for both the ‘presenilinase' and g-secretase
(refs 78–80). Establishing definitively that Ab accumulation
cleavages, or function as the active site of an intramembranous
triggers the hyperphosphorylation of tau, which precedes the
aspartyl protease by cleaving the Ab
assembly of these molecules into PHF13, must await the production
40–41 and Ab42–43 peptide bonds
within C99 and C83 to generate Ab
of neurofibrillary tangles in transgenic mice that overexpress both
40 and Ab42 and p340 and p342.
Two additional findings are consistent with the second mechanism.
mutant APP and human tau. Although tangles have not been
First, mutating either of the aspartates to glutamate again disrupts
described in existing APP mouse models81–83, crossing APP/
the presenilinase and g-secretase cleavages, indicating that the
presenilin doubly transgenic mice with mice that overexpress
conserved, charged glutamate residue cannot substitute for the
human tau bearing one of the tangle-promoting mutations might
aspartate65. Second, Ab can be generated from recombinantly
be expected to lead to a ‘plaque-plus-tangle' phenotype that closely
expressed C99 in vitro using microsomes containing wild-type but
resembles the AD state.
not Asp-mutant PS1 and at mildly acidic but not neutral pH65.
Although substantial evidence supports the Ab-mediated cyto-
Although these various findings suggest that the presenilins
pathological cascade summarized briefly in the preceding para-
represent the long-sought g-secretases, definitive proof will require
graph, many questions remain unanswered. First, what are the
reconstitution of Ab generation from pure C99 and pure wild-type
relative contributions of extracellular compared with intraneuronal
(but not Asp-mutant) PS1 in phospholipid vesicles, something that
Ab accumulation in initiating the neurotoxic response? Whereas
may be difficult to accomplish until other protein factors that are
immunohistochemistry shows abundant extracellular Ab in AD
believed to regulate presenilin fragments are discovered40. If pre-
brains, the same antibodies seem not to detect any intracellular
senilins are, indeed, g-secretases, they could effect the intra-
accumulation. Nevertheless, small amounts of Ab dimers or higher
membranous cleavage of other substrates, in particular the Notch
oligomers could well be accumulating intracellularly69, and their
proteins, which apparently undergo intramembranous proteolysis67
role in neuronal dysfunction will need to be established. Second, are
and which require the presenilins for their signalling function29,34. In
Ab fibrils the principal toxic moiety in the disease, or do they
this regard, mutation of just one of the PS1 transmembrane
instead represent a relatively inert aggregate, so that smaller assem-
aspartates to asparagine has been found to destroy the ability of
bly forms such as protofibrils84,85 or even diffusible dimers or other
human PS1 to rescue the lethal phenotype of the sel-12 mutation in
oligomers86 actually serve as the microglia-activating and neuron-
injuring species? In this context, are highly fibrillar ‘mature' plaques
Confirmation that presenilins are g-secretases would provide
simply a consequence of the local accumulation of Ab to high levels,
further support for the amyloid cascade hypothesis of AD. The most
allowing smaller, diffusable species of the peptide in equilibrium
common mutations causing familial (autosomal dominant) AD
with the fibrils to serve as the toxic moieties?
would therefore occur in the very protease that generates Ab, and
Another pressing question is whether apoptosis of neurons is
mutations in either the substrate or the protease would result in a
important for producing AD brain dysfunction. Although pre-
markedly accelerated and severe AD phenotype.
senilins (particularly mutant PS2) have been associated with
It is important to emphasize that each new gene implicated in
enhanced apoptosis in cell-culture studies87, whether and how
familial forms of AD will need to be similarly analysed to establish
presenilins mediate apoptosis in vivo will need to be considered in
its specific effect on the production, deposition or clearance of Ab,
light of the possibility that presenilin is an aspartyl protease that
in order to determine whether all genetic forms of the disorder
processes APP and probably other intramembranous substrates
involve an elevation of steady-state levels of Ab.
such as Notch65. Moreover, the ultrastructural appearance of the
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
plaque formation could be of potential therapeutic interest. Thesecond level at which selective vulnerability may act relates to the
Missense mutations in APP, PS1 and PS2 genes
observation that not all neurons and neurites in the vicinity ofmature, fibril-rich plaques undergo neurotoxic changes. The com-plex issue of what allows some neurons to succumb and others to
Altered proteolysis of APP
survive may be among the most difficult to resolve, as it has beenin many other selective neurodegenerative disorders of varyingaetiology.
Increased production of Aβ42
Notwithstanding the many unanswered questions about the
inflammatory and neurotoxic cascade of AD, studies of disease
Progressive accumulation and aggregation of Aβ42
progression in Down's syndrome and transgenic mouse models
in brain interstitial fluid
continue to support Ab accumulation as an early event in most orall forms of the syndrome. Interference with early steps in theprocess, such as Ab production or Ab assembly, may therefore
Deposition of aggregated Aβ42 as diffuse plaques
prove a more attractive therapeutic strategy than attempting to
(in association with proteoglycans and other
block the multiple downstream effects of the peptide and its many
Aggregation of Aβ
Predictions for pharmacological intervention
40 onto diffuse Aβ42 plaques
Accrual of certain plaque-associated
Despite our incomplete understanding of AD, sufficient progress in
proteins (for example, complement c1q)
delineating the disease cascade has been achieved to suggest severaldiscrete targets for treatment. These include: (1) inbititors of Abproduction (that is, small compounds that decrease but do not
'Inflammatory' response:
eliminate b- or g-secretase activity); (2) inhibitors of Ab oligo-
• Microglial activation and cytokine release
merization or fibrillization; (3) anti-inflammatory drugs that could
• Astrocytosis and acute-phase protein release
interfere with aspects of the microglial and astrocytic responses inthe brain; (4) antioxidants, free-radical scavengers, calcium-channelblockers and modulators of signal transduction that could protect
Progressive neuritic injury within amyloid
plaques and elswhere in the neuropil
neurons from the downstream effects of the accumulation of Aband its associated proteins; and (5) neurorestorative factors (forexample, neurotrophins and small compounds mimicking their
Disruption of neuronal metabolic and ionic
action; oestrogens) that could conceivably rescue synapses and cell
homeostasis; oxidative injury
bodies undergoing active injury. All of these approaches should bepursued, because success of one particular strategy cannot bepredicted and two or more approaches might ultimately be com-
bined to treat a patient, depending on the temporal stage in the
phosphatase activities Hyperphosphorylated
disorder. Current, largely symptomatic treatments aimed at
tau PHF formation
enhancing the levels of depleted neurotransmitters such asacetylcholine may still be used, even if more specific treatmentsaimed at early steps in the disease are forthcoming. Based on the
Widespread neuronal/neuritic dysfunction
and death in hippocampus and cerebral
emerging importance of Ab, g-secretase inhibitors or other Ab-
cortex with progressive neurotransmitter deficits
lowering compounds may be the first agents to reach clinical trials,although it is possible that partial inhibition of this highly unusualprotease may have serious side effects. Although downregulating
Notch and APP processing could be considered as dangerousconsequences of a g-secretase inhibitor, the therapeutic goal is toinduce partial (perhaps 30–40%) inhibition of the protease, just as
Figure 4 A hypothetical sequence of the pathogenetic steps of familial forms of
has been safely accomplished for another vital enzyme, 3-hydroxy-
3-methylglutaryl coenzyme A (HMG CoA) reductase, in chronicallylowering cholesterol.
In the future, it is likely that individuals reaching their fifties or
many tangle-bearing neurons present in the AD brain suggests a
beyond will be offered a specific risk-assessment profile to deter-
gradual dysfunction and slow death of the affected neurons rather
mine their likelihood of developing AD. Such an assessment,
than a sudden, apoptotic loss. Tangle-bearing neurons often show
perhaps modelled on that now widely used to judge the risk of
relatively well preserved organelle structure by electron microscopy,
serious atherosclerotic disease, would include inquiry about a
with intact ER, nuclear membranes and mitochondria, which
positive family history, identification of specific predisposing
suggests that the cells are chronically dysfunctional but not near
genetic factors, structural and functional brain imaging to detect
apoptosis. Yet another unresolved question concerns the selective
evidence of presymptomatic lesions, and measurement of Ab42, tau
vulnerability of neuronal populations in AD. Local and regional
and other markers of the neuropathology in the spinal fluid and
differences in the pathogenic process arise on at least two broad
perhaps (in the case of Ab) even in the blood. Based on further
levels. First, Ab42 can accumulate chronically in some brain regions
epidemiological experience with such assessment measures in large
(for example, cerebellum, striatum and thalamus) with little or no
populations of elderly and AD subjects, it should be possible to
evolution to amyloid fibrils and their associated neuritic and glial
estimate—first crudely and later more accurately—the likelihood
cytopathology. At this level, regionally specific factors (for example,
that an individual will develop AD. If this can be accomplished, then
pro- or anti-aggregating proteins) may exist that enable Ab to
those at particularly high risk could be offered preventative treat-
proceed into aggregated forms or prevent it from doing so. Anti-
ment with one or more of the agents contemplated in the previous
aggregating factors that are present in regions spared from mature
paragraph. Although the achievement of an integrated diagnostic
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
and therapeutic approach to this complex and tragic disorder may
38. Podlisny, M. B. et al. Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and
still seem remote, the current rate of scientific progress indicates
299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue.
Neurobiol. Dis. 3, 325–337 (1997).
that some level of practical success may come sooner than one might
39. Li, X. & Greenwald, I. Additional evidence for an eight-transmembrane-domain topology for
think. If so, the knowledge gained from mechanistic and therapeutic
Caenorhabditis elegans and human presenilins. Proc. Natl Acad. Sci. USA 95, 7109–7114 (1998).
40. Thinakaran, G. et al. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by
research on AD should greatly facilitate efforts to understand
competition for limiting cellular factors. J. Biol. Chem. 272, 28415–28422 (1997).
and treat other brain disorders, for example, Parkinson's and
41. Steiner, H. et al. Expression of Alzheimer's disease-associated presenilin-1 is controlled by
proteolytic degradation and complex formation. J. Biol. Chem. 273, 32322–32331 (1998).
Huntington's diseases, that may also involve abnormal protein
42. Capell, A. et al. The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form
heterodimers and occur as a 100-150-kDa molecular mass complex. J. Biol. Chem. 273, 3205–3211
(1998).
Dennis J. Selkoe is at the Center for Neurologic Diseases, Harvard Medical School,
43. Zhang, J. et al. Subcellular distribution and turnover of presenilins in transfected cells. J. Biol. Chem.
Brigham and Women's Hospital, Boston, Massachusetts 02115, USA.
273, 12436–12442 (1998).
44. Scheuner, D. et al. Secreted amyloid b-protein similar to that in the senile plaques of Alzheimer's
Dickson, D. W. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 56, 321–339 (1997).
disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's
Yamaguchi, H., Nakazato, Y., Hirai, S., Shoji, M. & Harigaya, Y. Electron micrograph of diffuse
disease. Nature Med. 2, 864–870 (1996).
plaques: initial stage of senile plaque formation in the Alzheimer brain. Am. J. Pathol. 135, 593–597
45. Tomita, T. et al. Molecular dissection of domains in mutant presenilin 2 that mediate overproduction
of amyloidogenic forms of amyloid beta peptides. Inability of truncated forms of PS2 with familial
Goedert, M., Trojanowski, J. Q. & Lee, V. M.-Y. in The Molecular and Genetic Basis of Neurological
Alzheimer's disease mutation to increase secretion of Abeta42. J. Biol. Chem. 273, 21153–21160
Disease, 2nd edn (eds Rosenberg, R. N., Prusiner, S. B., DiMauro, S. & Barchi, R. L.) 613–627
(Butterworth-Heinemann, Boston, 1996).
46. Selkoe, D. J. Alzheimer's disease: genotypes, phenotype, and treatments. Science 275, 630–631 (1997).
Strittmatter, W. J. et al. Apolipoprotein E: high-avidity binding to b-amyloid and increased
47. Lemere, C. A. et al. Sequence of deposition of heterogeneous amyloid b-peptides and Apo E in Down
frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl Acad. Sci. USA 90,
syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32
1977–1981 (1993).
Saunders, A. M. et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and
48. Mann, D. M. et al. Microglial cells and amyloid beta protein (A beta) deposition; association with A
sporadic Alzheimer's disease. Neurology 43, 1467–1472 (1993).
beta 40-containing plaques. Acta Neuropathol. (Berl.) 90, 472–477 (1995).
Sherrington, R. et al. Cloning of a novel gene bearing missense mutations in early onset familial
49. Levy, E. et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage,
Alzheimer disease. Nature 375, 754–760 (1995).
Dutch-type. Science 248, 1124–1126 (1990).
Levy-Lahad, E. et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science
50. Hendriks, L. et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of
269, 973–977 (1995).
the b-amyloid precursor protein gene. Nature Genet. 1, 218–221 (1992).
Rogaev, E. I. et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on
51. Biere, A. L. et al. Co-expression of b-amyloid precursor protein (bAPP) and apolipoprotein E in cell
chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376, 775–778 (1995).
culture: analysis of bAPP processing. Neurobiol. Dis. 2, 177–187 (1995).
Blacker, D. et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nature
52. Schmechel, D. E. et al. Increased amyloid b-peptide deposition in cerebral cortex as a consequence of
Genet. 19, 357–360 (1998).
apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl Acad. Sci. USA 90, 9649–9653
10. Spillantini, M. G. et al. Mutation in the tau gene in familial multiple system tauopathy with presenile
dementia. Proc. Natl Acad. Sci. USA 95, 7737–7741 (1998).
53. Rebeck, G. W., Reiter, J. S., Strickland, D. K. & Hyman, B. T. Apolipoprotein E in sporadic
11. Hutton, M. et al. Association of missense and 59-splice-site mutations in tau with the inherited
Alzheimer's disease: allelic variation and receptor interactions. Neuron 11, 575–580 (1993).
FTDP-17. Nature 393, 702–705 (1998).
54. Ma, J., Yee, A., Brewer, H. B. Jr, Das, S. & Potter, H. The amyloid-associated proteins
12. Poorkaj, P. et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol.
a1-antichymotrypsin and apolipoprotein E promote the assembly of the Alzheimer b-protein into
43, 815–825 (1998).
filaments. Nature 372, 92–94 (1994).
13. Goedert, M. Tau mutations cause frontotemporal dementias. Neuron 21, 955–958 (1998).
55. Evans, K. C., Berger, E. P., Cho, C.-G., Weisgraber, K. H. & Lansbury, P. T. Jr Apolipoprotein E is a
14. Kang, J. et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface
kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis
receptor. Nature 325, 733–736 (1987).
and treatment of Alzheimer disease. Proc. Natl Acad. Sci. USA 92, 763–767 (1995).
15. Glenner, G. G. & Wong, C. W. Alzheimer's disease and Down's syndrome: sharing of a unique
56. Bales, K. R. et al. Lack of apolipoprotein E dramatically reduces amyloid b-peptide deposition.
cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135 (1984).
Nature Genet. 17, 263–264 (1997).
16. Selkoe, D. J. Cell biology of the amyloid b-protein precursor and the mechanism of Alzheimer's
57. Hardy, J. The Alzheimer family of diseases: many etiologies, one pathogenesis? Proc. Natl Acad. Sci.
disease. Annu. Rev. Cell Biol. 10, 373–403 (1994).
USA 94, 2095–2097 (1997).
17. Zheng, H. et al. b-amyloid precursor protein-deficient mice show reactive gliosis and decreased
58. Holcomb, L. et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant
locomotor activity. Cell 81, 525–531 (1995).
amyloid precursor protein and presenilin 1 transgenes. Nature Med. 4, 97–100 (1998).
18. Perez, R. G., Zheng, H., Van der Ploeg, L. H. & Koo, E. H. The beta-amyloid precursor protein of
59. Lemere, C. A. et al. The E280A presenilin 1 Alzheimer mutation produces increased Ab42 deposition
Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J. Neurosci. 17,
and severe cerebellar pathology. Nature Med. 2, 1146–1148 (1996).
9407–9414 (1997).
60. Mann, D. M. A. et al. Amyloid beta protein (A-beta) deposition in chromosome 14-linked
19. Wasco, W. et al. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer
Alzheimer's disease—predominance of A-beta (42(43)). Ann. Neurol. 40, 149–156 (1996).
disease-associated amyloid b-protein precursor. Proc. Natl Acad. Sci. USA 89, 10758–10762 (1992).
61. Thinakaran, G. et al. Stable association of presenilin derivatives and absence of presenilin
20. Slunt, H. H. et al. Expression of a ubiquitous, cross-reactive homologue of the mouse b-amyloid
interactions with APP. Neurobiol. Dis. 4, 438–453 (1998).
precursor protein (APP). J. Biol. Chem. 269, 2637–2644 (1994).
62. Xia, W. et al. Presenilin 1 regulates the processing APP C-terminal fragments and the generation of
21. Sisodia, S. S. b-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl Acad.
amyloid b-protein in ER and Golgi. Biochemistry 37, 16465–16471 (1998).
Sci. USA 89, 6075–6079 (1992).
63. Weidemann, A. et al. Formation of stable complexes between two Alzheimer's disease gene products:
22. Black, R. A. et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-a from cells.
presenilin-2 and b-amyloid precursor protein. Nature Med. 3, 328–332 (1997).
Nature 385, 729–733 (1997).
64. Xia, W., Zhang, J., Perez, R., Koo, E. H. & Selkoe, D. J. Interaction between amyloid precursor protein
23. Buxbaum, J. D. et al. Evidence that tumor necrosis factor alpha converting enzyme is involved in
and presenilins in mammalian cells: implications for the pathogenesis of Alzheimer's disease. Proc.
regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273,
Natl Acad. Sci. USA 94, 8208–8213 (1997).
65. Wolfe, M. S. et al. Two transmembrane aspartates in presenilin-1 required for presenilin endo-
24. Haass, C. et al. Amyloid b-peptide is produced by cultured cells during normal metabolism. Nature
proteolysis and g-secretase activity. Nature 398, 513–517 (1999).
359, 322–325 (1992).
66. De Strooper, B. et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor
25. Shoji, M. et al. Production of the Alzheimer amyloid b protein by normal proteolytic processing.
protein. Nature 391, 387–390 (1998).
Science 258, 126–129 (1992).
67. Schroeter, E. H., Kisslinger, J. A. & Kopan, R. Notch-1 signalling requires ligand-induced proteolytic
26. Seubert, P. et al. Isolation and quantitation of soluble Alzheimer's b-peptide from biological fluids.
release of intracellular domain. Nature 393, 382–386 (1998).
Nature 359, 325–327 (1992).
68. Brockhaus, M. et al. Caspase-mediated cleavage is not required for the activity of presenilins in
27. Busciglio, J., Gabuzda, D. H., Matsudaira, P. & Yankner, B. A. Generation of b-amyloid in the
amyloidogenesis and NOTCH signaling. NeuroReport 9, 1481–1486 (1998).
secretory pathway in neuronal and nonneuronal cells. Proc. Natl Acad. Sci. USA 90, 2092–2096
69. Skovronsky, D. M., Doms, R. W. & Lee, V. M.-Y. Detection of a novel intraneuronal pool of insoluble
amyloid b protein that accumulates with time in culture. J. Cell Biol. 141, 1031–1039 (1998).
28. Seubert, P. et al. Secretion of b-amyloid precursor protein cleaved at the amino-terminus of
70. Eikelenboom, P., Zhan, S. S., van Gool, W. A. & Allsop, D. Inflammatory mechanisms in Alzheimer's
the b-amyloid peptide. Nature 361, 260–263 (1993).
disease. Trends Pharmacol. Sci. 15, 447–450 (1994).
29. Levitan, D. & Greenwald, I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis
71. McGeer, P. L. & McGeer, E. G. The inflammatory response system of brain: implications for therapy
elegans S182 Alzheimer's disease gene. Nature 377, 351–354 (1995).
of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 21, 195–218 (1995).
30. Levitan, D. et al. Assessment of normal and mutant human presenilin function in Caenorhabditis
72. Rogers, J. et al. Inflammation and Alzheimer's disease pathogenesis. Neurobiol. Aging 17, 681–686
elegans. Proc. Natl Acad. Sci. USA 93, 14940–14944 (1996).
31. Baumeister, R. et al. Human presenilin-1, but not familial Alzheimer's disease (FAD) mutants,
73. Rogers, J. et al. Complement activation by b-amyloid in Alzheimer disease. Proc. Natl Acad. Sci. USA
facilitate Caenorhabditis elegans notch signalling independently of proteolytic processing. Genes
89, 10016–10020 (1992).
Function 1, 149–159 (1997).
74. Itagaki, S., Akiyama, H., Saito, H. & McGeer, P. L. Ultrastructural localization of complement
32. Zhou, J. et al. Presenilin 1 interaction in the brain with a novel member of the Armadillo family.
membrane attack complex (MAC)-like immunoreactivity in brains of patients with Alzheimer's
NeuroReport 8, 2085–2090 (1997).
disease. Brain Res. 645, 78–84 (1994).
33. Yu, G. et al. The presenilin 1 protein is a component of a high molecular weight intracellular complex
75. Behl, C., Davis, J. B., Lesley, R. & Schubert, D. Hydrogen peroxide mediates amyloid b protein
that contains beta-catenin. J. Biol. Chem. 273, 16470–16475 (1998).
toxicity. Cell 77, 817–827 (1994).
34. Wong, P. et al. Presenilin 1 is required for Notch 1 and D111 expression in the paraxial mesoderm.
76. Harris, M. E., Hensley, K., Butterfield, D. A., Leedle, R. A. & Carney, J. M. Direct evidence of oxidative
Nature 397, 288 (1997).
injury produced by the Alzheimer's beta-amyloid peptide (1-40) in cultured hippocampal neurons.
35. Shen, J. et al. Skeletal and CNS defects in presenilin-1 deficient mice. Cell 89, 629–639 (1997).
Exp. Neurol. 131, 193–202 (1995).
36. Qian, S. et al. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic
77. Sano, M. et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's
lethality and elevates Abeta1-42/43 expression. Neuron 20, 611–617 (1998).
disease. The Alzheimer's Disease Cooperative Study. N. Engl. J. Med. 336, 1216–1222 (1997).
37. Thinakaran, G. et al. Endoprotreolysis of presenilin 1 and accumulation of processed derivatives in
78. Mattson, M. P. et al. b-Amyloid peptides destabilize calcium homeostasis and render human cortical
vivo. Neuron 17, 181–190 (1996).
neurons vulnerable to excitotoxicity. J. Neurosci. 12, 379–389 (1992).
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
79. Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. Neurodegeneration
90. Iwatsubo, T. et al. Visualization of Ab42(43) and Ab40 in senile plaques with end-specific Ab
induced by b-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13, 1676–1687
monoclonals: evidence that an initially deposited species is Ab42(43). Neuron 13, 45–53 (1995).
91. Jarrett, J. T., Berger, E. P. & Lansbury, P. T. Jr The carboxy terminus of the beta amyloid protein is
80. Lorenzo, A. & Yankner, B. b-amyloid neurotoxicity requires fibril formation and is inhibited by
critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's
Congo red. Proc. Natl Acad. Sci. USA 91, 12243–12247 (1994).
disease. Biochemistry 32, 4693–4697 (1993).
81. Games, D. et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F
92. Yamazaki, T., Koo, E. H. & Selkoe, D. J. Trafficking of cell-surface amyloid b-protein precursor. II.
b-amyloid precursor protein. Nature 373, 523–527 (1995).
Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J. Cell Sci. 109,
82. Hsiao, K. et al. Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice.
999–1008 (1996).
Science 274, 99–102 (1996).
93. Yamazaki, T., Selkoe, D. J. & Koo, E. H. Trafficking of cell surface b-amyloid precursor protein:
83. Sturchler-Pierrat, C. et al. Two amyloid precursor protein transgenic mouse models with Alzheimer
retrograde and transcytotic transport in cultured neurons. J. Cell Biol. 129, 431–442 (1995).
disease-like pathology. Proc. Natl Acad. Sci. USA 94, 13287–13292 (1997).
94. Koo, E. H. & Squazzo, S. Evidence that production and release of amyloid b-protein involves the
84. Harper, J. D., Wong, S. S., Lieber, C. M. & Lansbury, P. T. Jr Observation of metastable Ab amyloid
endocytic pathway. J. Biol. Chem. 269, 17386–17389 (1994).
protofibrils by atomic force microscopy. Chem. Biol. 4, 119–125 (1997).
95. Wilde-Bode, C. et al. Intracellular generation and accumulation of amyloid b-peptide terminating at
85. Walsh, D. M. et al. Amyloid b-protein fibrillogenesis: detection of a protofibrillar intermediate.
amino acid 42. J. Biol. Chem. 272, 16085–16088 (1997).
J. Biol. Chem. 272, 22364–22374 (1997).
96. Cook, D. G. et al. Alzheimer's Ab (1–42) is generated in the endoplasmic reticulum/intermediate
86. Lambert, M. P. et al. Diffusible, nonfribrillar ligands derived from Ab1–42 are potent central nervous
compartment of NT2N cells. Nature Med. 3, 1021–1023 (1997).
system neurotoxins. Proc. Natl Acad. Sci. USA 95, 6448–6453 (1998).
97. Chyung, A. S. C., Greenberg, B. D., Cook, D. G., Doms, R. W. & Lee, V. M.-Y. Novel b-secretase
87. Wolozin, B. et al. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an
cleavage of b-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment
Alzheimer mutation. Science 274, 1710–1713 (1996).
of NT2N cells. J. Cell Biol. 138, 671–680 (1997).
88. Miller, D. L., Papayannopoulos, I. A., Styles, J. & Bobin, S. A. Peptide compositions of the
98. Haass, C. et al. The Swedish mutation causes early-onset Alzheimer's disease by b-secretase cleavage
cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch. Biochem.
within the secretory pathway. Nature Med. 1, 1291–1296 (1995).
Biophys. 301, 41–52 (1993).
99. Hartmann, T. et al. Distinct sites of intracellular production for Alzheimer's disease Ab-40/42
89. Roher, A. E. et al. b-amyloid-(1–42) is a major component of cerebrovascular amyloid deposits:
amyloid peptides. Nature Med. 3, 1016–1020 (1997).
implications for the pathology of Alzheimer's disease. Proc. Natl Acad. Sci. USA 90, 10836–10840
100. Lee, S. J. et al. A detergent-insoluble membrane compartment contains Ab in vivo. Nature Med. 4,
730–734 (1998).
1999 Macmillan Magazines Ltd
NATURE VOL 399 SUPP 24 JUNE 1999 www.nature.com
Source: http://azolla.fc.ul.pt/aulas/BiologiaCelular/docs/Alzheimer.pdf
Calendario d g ite tturistiche ee d d ei ssoggiorni mare - montagna Festa del Tulipano a Castiglione del Lago dall' 11 al 12 aprile € 190 Il punto Terme di Monticelli dal 4 al 16 maggio €740 Responsabile del turismo: sig.ra Mida: 338-4932066 Le mie esperienze in Uniauser
J Nanopart Res (2013) 15:1879DOI 10.1007/s11051-013-1879-8 Improved photodynamic action of nanoparticles loadedwith indium (III) phthalocyanine on MCF-7breast cancer cells Carlos Augusto Zanoni Souto • Kle´sia Pirola Madeira • Daniel Rettori •Mariana Ozello Baratti • Letı´cia Batista Azevedo Rangel • Daniel Razzo •Andre´ Romero da Silva Received: 1 April 2013 / Accepted: 16 July 2013Ó Springer Science+Business Media Dordrecht 2013



