Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Slide
Survey Results & analysis for
Global survey of models of
preclinical TB drug testing
This report contains a detailed statistical analysis of the results to the survey titled Global survey of models of preclinical TB drug testing . The results analysis includes answers from all respondents who took the survey (30 completed responses were received to the survey up to this time)
Draft survey: updated April 18, 2008Please address concerns or questions to Mary Ann De
[email protected]
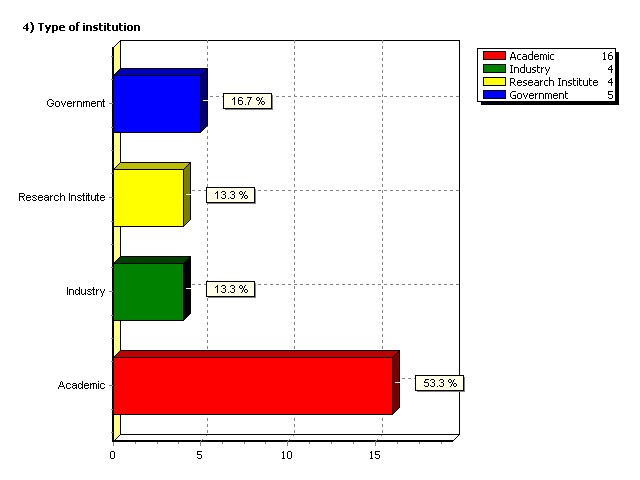
Type of institution
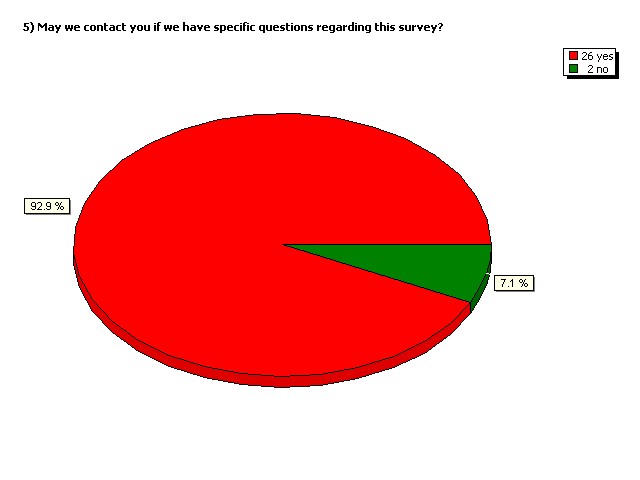
May we contact you if we have
specific questions regarding
this survey?
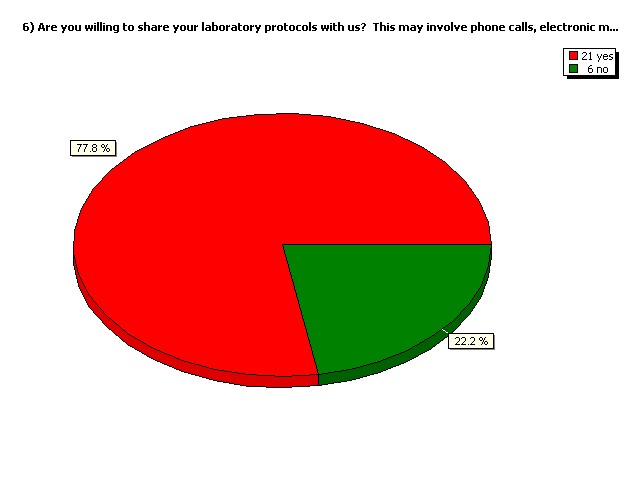
Are you willing to share your
laboratory protocols with us?
This may involve phone calls,
electronic mail etc.
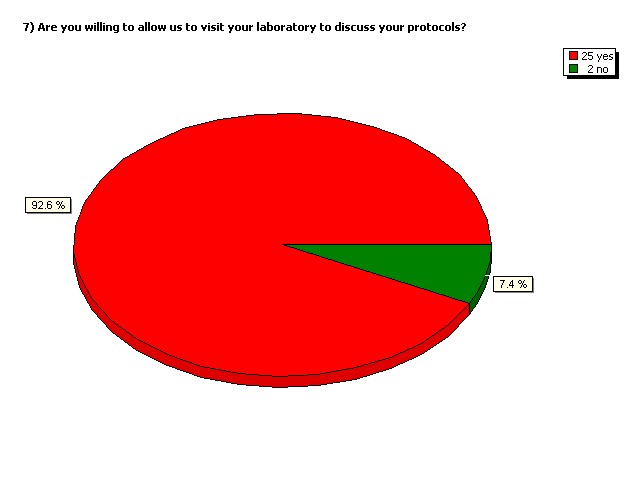
Are you willing to allow us to visit
your laboratory to discuss your
protocols?
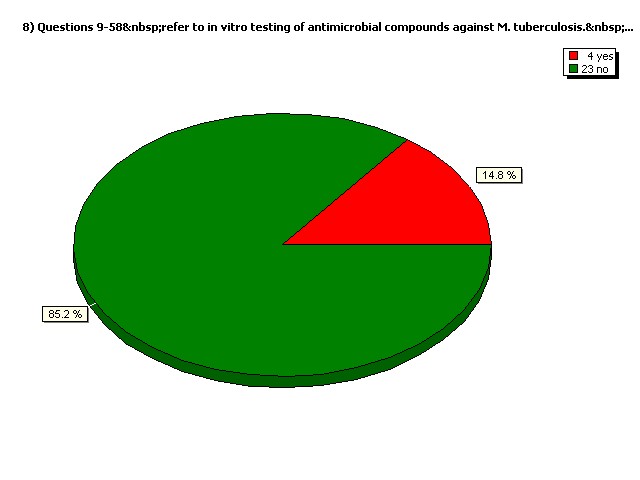
Do you perform in vivo (animal model) testing exclusively?
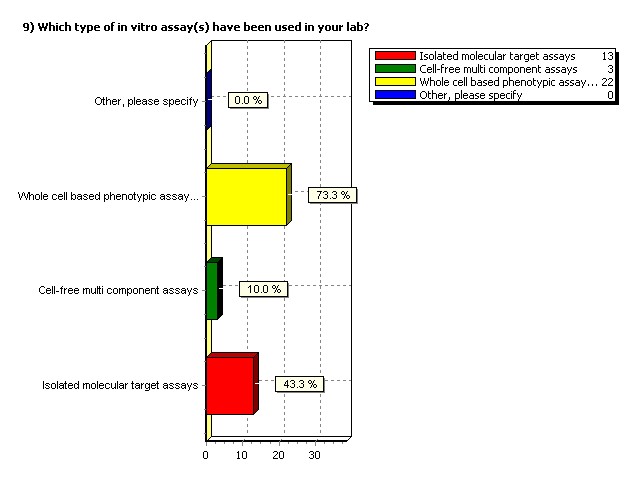
Which type of in vitro assay(s)
have been used in your lab?
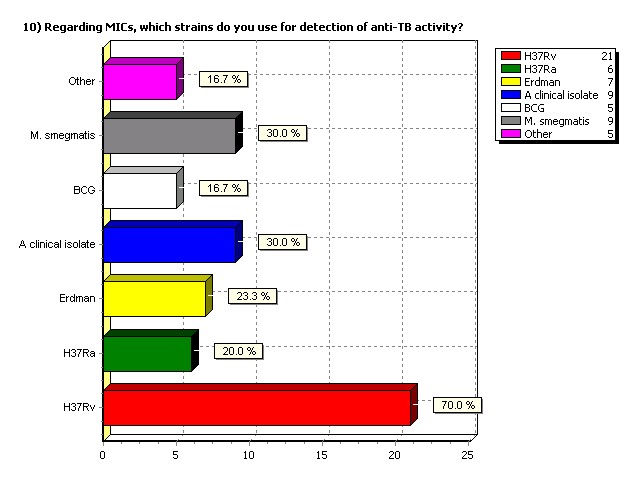
Regarding MICs, which strains do
you use for detection of anti-TB
activity?
Other responses: regarding strains
We do not perform MIC testing
W-Beijing; CDC1551; H37Rv ATCC 25618
MAC STRAINS and fast growing mycobacteri
Mono, poly and multi drug resistant stra
Epidemiological representative strains
Drug resistant strains
Any significant differences in activity between the surrogate strain and virulent M. tuberculosis?
Yes. Strain Ra has been compared to strain Rv and difference can be found in sensitivity to new compounds. One possibitlity is that the deletions in Ra could code for a protein that activates a drug, and if deleted Ra would be resistant. Another alternative is the example of the erm methylase. If deleted in Ra the strain would now be sensitive to macrolides.
Yes, BCG is more susceptible to nitro-imidazoles versus our standard H37Rv
We use virulent strain of MTB (H37Rv) for standard MIC testing.When BCG is used for large scale screening we observe more than 90% overlap with H37Rv.
The correlation is good between BCG and TB. Not so with Smeg and TB.
Differences between H37Rv and H37Ra
Source or history of the strain used.
ATCC, CSU and Pasteur Institute
M. smegmatis mc2155 from Dr. Jacobs' lab
D.Y. Young, ex Imperial College London
M. bovis BCG- Pasteur and above mentioned clinical strains
a. H37Rv ATCC27294 b. serious drug resistant clinical isolates dependent on the natural product sources
H37Rv lab strain, and BCG-Russia
H37Rv is from ATCC Other clinical strains used include HN878 and CDC1551
mc2155 (JAcobs strain)
Strains were collected from the local hospitals
Strains obtained from Tuberculosis Research Center Chennai, INDIA
Ra, original source unknown.
Erdman from the ATCC H37Rv from Trudeau Institute
H37Rv from ATCC (# 27294) BCG from ATCC (#35734)
ATCC TRC, Chennai - clinical isolates
H37Rv and Erdman: ATCC Clinical SNP cluster representatives: David Alland, UMDNJ H37Rvlux AB: constructed in house Moxifloxacinresistant: constructed in house
Is your strain naturally drug resistant or
marked with an antibiotic selection marker?
Please elaborate on the nature of the drug resistance.
- Spontaneous mutants - well characterized clinical isolates
a. H37Rv - No b. clinical isolates - Yes, they are naturally drug resistant strains
MDR, XDR, Monoresitant strains
Not for standard MIC testing. Lead compounds are tested against appropriate mono, poly and/or multi resistant strains (clinical strains or strains derived in vitro after drug selection)
Except for the clinical isolates, the standard lab strains are not resistant or marked.
Various strains with different resistant patterns
H37Rv lux AB: kanamycin marker monoresistant strains to rifampin, kanamycin, streptomycin, etc. are isogenic to H37Rv
Regarding the culture medium used to grow the mycobacterial strain for inoculation into the MIC assays, please elaborate on medium used
(including pH, carbon source, method of sterilization, culture time, culture vessels, growth phase when harvested and supplements added)
for both inoculum preparation and the anti-TB assay. If this is different for the inoculum preparation and the anti-TB testing, indicate why it is different.
Std Middlebrook 7H9 O
y ine-alanine salt (GAS) medium
upple ented with T
80, pH 6.6 Bacto-
t Casitone, citric acid, L-alanine, glyc
believed to be the m
lave at 121C for 30 m
g atis inoculum was
w cultured in a test t
2 day in GAS supplemented with T
80 For MIC determination, M. smeg
g atis are cultured in 96-
w l plate overnight to log
- Culture on 7H11 or 7H10 - Als
A o 7H11 pH (with HCl prior to autoclaving) 5.9 for pza
Middlebrook 7H9 broth
oth f or both inoc
ulation and MIC assay -
auto lave, culture in erlenmayer
plask upto A600 0.5 (
u e 7H9, pH6.6, glyc
y erol as carbon source, autoc
lave sterilization, t
and ADC are added af
e added a ter autoc
auto laving, culture time var
with experiment, cultur
y ene bottles on a roller
y erol + 0.05% Tween
80. Either filter ster
autoclaved according to
e e looking at Fe-
e w ici is filter-sterilized: G
liter: 0.3 g of Bacto
t Casitone (Difco)
o citric acid, 1.0 g of
acid, 1.0 g o L-alanine, 1.2 g of
hloride hexahydr
y ate, 0.6 g of potass
chloride, 1.80 ml of
y oxide, and 10.0 m
ide, and 10.0 l of gl
w added to 0.
added to 0 05%. GAS
above with 0.05 g of
above with 0.05 g o fe
Plates: Mueller-Hinton, hyg
oth containing tween
Regarding the culture medium used to grow the mycobacterial strain for inoculation into the MIC assays, please elaborate on medium used
(including pH, carbon source, method of sterilization, culture time, culture vessels, growth phase when harvested and supplements added)
for both inoculum preparation and the anti-TB assay. If this is different for the inoculum preparation and the anti-TB testing, indicate why it is different.
y available Middlebrook 7H 11
. or inoculam we ar
ence o betalactam
a e inhibitors. W
it appear that c
that ells may not s
t t active growth (
w ld be sensitive) immediately and thus
uch higher than ex
ediu , grown as biof
y erol alanine salts pH6.6 and/or
pH6.6 and/ 7H9-OADC (pH 6.6) autoclaved and OADC f
in 96 w ll U-bottomed plates
Medium: 7H9-ADS (albumin 5%, glucose 2%
and NaC 0.8% final concentration) + gl
y erol 0.5%+ 0.05% Tween
80 pH: 6.6 Method o
80 pH: 6.6 Method f
z ion: filtration through 0.22µM mem
ane ilter Culture time: 5 days
y Culture vessel: microtiter plates (96-
w ll-plates) Phase of
owing cells that wer
that w e concentrated b
y entrifugation (at an O
80oC prior to use. Dilution
( or M. tuberculosis): method o
t ilization: autocalave; pH: 6.8; culture tim
upple ents added: 10%
en ( or M. tuberculosis): method of
alave; culture time: 4-6 wee
(for M. tuberculosis): m
d o sterilization: autocalave; pH: 7; c
y , supplements added: 1
Broth (for M. smeg
t ilization: autoc
on: auto alave; pH: 7; culture time: 2 da
Middlebrook 7H9 ADC; autoc
7H9 ADC; aut laving; 7day R
ay in 96 or 384 we
ting inoculum from
o f rozen seedlot wh
ting ell number 5.5 x 10 5/m
l. Seedlot prepared f
e cultures grown in r
oller bottles, whic
5 colonies pooled f
7H12 broth at various pHs
7H12 medium, pH 6.8, c
e: late log, Supplements
ent added: bovine s
ing do not contain this supplem
Macrophage assays used:
e lines in experimental developm
e e H37Rv. There c
an be uite big dif
en es in MIC between in
vitro and macrophage det
y , its clear that som
y gainst Mtb in macrophages
t MTb in vitro.
phage ells in vitro assays
e ing J774A.1 & T
o intracellular assays
u ed :Mtuberculos
ee the difference in reg
in vitro MIC and ex vivo activity,this
t related to clas
ve observed discrepancies when
vo data testing a w
ing a w de variety of
d ug classes we obs
wing: three out of
vivo active compounds did not s
y in the macrophage m
ophage odel. Reas
on is not clear and has
and ha not be pu
e pu sued, could be due t
uld be due t lack of
ay is questionable (streptom
amikacin are poor
tive in the assay)
ophage or in vitro assays
y for M. tuberculosis.
A CC)and Murine BMDM (primary c
y ells) H37Rv (AT
A CC 27294) Many c
y ompounds that have pote
dont have good ac
have good tiv ity in
ay - not surprising due to intracellular PK influences. Lik
ew se, those that ar
ood in acrophage ar
v for in vivo testing and not
ing and not ed as
ed a exclusion criteria.
774 w th M. tuberculosis Erdman, s
What is your default stock concentration for new compounds?
Depends on solubility
50-100mg/ml 100mg/ml favored
Depends on solubility. We try 50mM in DMSO
I don't understand the question
from 50microg/ml to lower concentration
I don't use new compound
10 mg/ml or 10 uM
5 mM in 90% DMSO (Di Methyl Sulfoxide)
2 mM for the stocks and 25 microM in the plate.
12.8 mM or 12.8 or 10 mg/ml
Do you prepare all compounds fresh each day or do you use frozen stocks?
Frozen stocks.
- Strongly favor stocks
We prepare fridge stocks from frozen stocks
Frozen unless there are indications of instability
I don't use frozen stock
No. We do make stock of the compounds and working stocks were used for further assays
Both depending on stability data.
Prepare fresh for every experiment
We prepare stock solutions once a week.
frozen stocks stored at -80oC
Compounds are stored at 4oC prior to use, both fresh and old stock used. Integrity of the compounds is verified by LC-MS on a regular basis
We use room temperature stocks.
frozen stocks but not stored for longer than 6 months
If using frozen stocks of compounds do you make aliquots
and use each one once or do you re-use the same frozen stocks?
Re-use the same frozen stocks
Re-use - provided from assay screening group in 96-well plates.
We re-use frozen stocks up to 3-4 times
Same stock unless there are indications of instability. Same stock is not thawed more than 10 times
Yes aliquots are made
Make aliquotes.
aliquotes, cmpds are never refrozen.
Working stocks aliquots are preapred and my be used more than once (multiple freeze-thaws)
Reuse the same frozen stock unless visible precipitation observed.The activity of a compound is always confirmed with a secong, independently prepared stock.
We make aliquots.
Frozen stocks are not used
If in microplates we re-use; if in tubes, we prepare aliquots
Do you filter sterilize stocks made in aqueous and/or organic solvents?
no (we perform enyzmatic assays)
we filter running solutions before use
Aqeous - yes. DMSO - no
Stocks are made under sterile conditions.
Only on aoccasion, e.g. betalactams in aqueous solution and filter sterilized.
Yes for aqueous. No for orgainc
We do filter sterilize stocks to be used for in vitro studies.
no, they are made up in DMSO (dimethylsulfoxide)
We dissolve the compounds in 100% DMSO and then make up the volume with 10% filter sterilized water. We do not filter sterilize the 90% DMSO stocks
We don't filter stocks.
Filter water-soliuable compounds
yes for aqueous, not for organic
Other than water, what other solvents do you use?
Please specify alternative solvents and for which compounds they are used
DMSO used for all compounds.
DMSO, ethanol, acetonitrile
DMSO (most of our compounds), sometimes ethanol
I don't use solvents
DMSO as a general solvent for most compounds
Methanol for rifampicin
ethanol, DMSO, PEG, cyclodextrin micelle (CM-2), carboxymethylcellulose
DMSO (dimethylsulfoxide) for nearly all our compounds we test
All compounds are dissolved in 90% DMSO to make the compound stocks. We do not use any other aqueous solvents.
We use DMSO for all the compounds which are not soluble in water.
DMSO is default solvent
What is the highest final concentration of DMSO in the assay plate that you use?
Did you do any tests to determine the highest concentration of DMSO used in the
plate without affecting the growth/viability of M. tuberculosis?
spectrophotometric assay 3% (DMSO up to 4.5% does not affect the enzyme activity. Higher concentrations were not tested.) radiometric assay 3%
1% We tested DMSO levels, and found 5% to be the upper limit.
Usually not above 1% although for some really inactives we have had to use 4%
we use DMSO to dissolve the compound and for dilution we use water.
1% and tested for each new assay.
Highest final DMSO concentration used is
The highest final concentration of DMSO in the assay plate is 1.25%. The inhibitory effect of different concentrations of DMSO against M. tuberculosis were tested many times.
Yes, see our publications
1 percent Have previously determined that M. tuberculosis can tolerate up to 1.5% DMSO
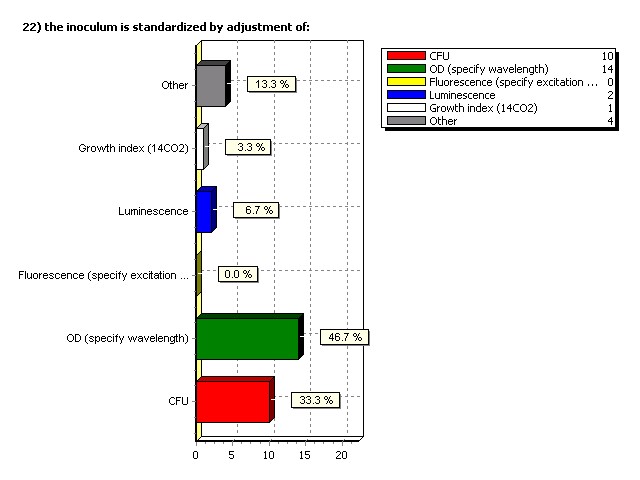
Inoculum is standardized by adjustment of:
Additional comments on inoculum
we measure purified enzyme activity
dependent on the test methods
McFarland using Densimat
Frozen seedlot with fixed inoculum
What is the target CFU per ml at the beginning of the incubation?*
What is the target CFU per ml at the beginning of the incubation?
about 2x 10 to the 5
10 000 cells per well
5 exp 4 CFUper well
5.5 x 10 5 cfu/ml
*Note: this question may not have been clear
Assay format includes
Additional comments on in vitro screening
for radiometric assay; 25 ul, in Eppis
CLSI Agar Dilution Method
20ml, metal closure
grown in 25 ml medium in 250 ml bottle
16mm tube, screw cap
15ml conical vial, 2.5ml broth, loosecap
Tube MIC:14 mL snap cap,final volume 1ml
tubes of 10 ml, 10 cm height, 1cm diameter
Bactec 460 12B vials
Total assay volume in each well or tube
spectro - 200 ul, r
adiometric - 25 ul
w ll plate: 200 µL, tube : 1m
plate: 200 µL, tube :
Are plates or tubes sealed? If so, how is this done?
a plate sealers.
Lids on but not s
on but not ealed, enc
ealed, en losed in sealed box to incubate.
Seal with breathable sealing tape (sheet)
with lid, not airtight seal
. hen closed in ziplock bags f rom
Y , tightening the c
Plates are not sealed. W
to limit evaporation.
no, cap is f itted loosely
sealed in zip lock bag for
Plates are not sealed
ith plate sealer.
Plates are in CO2 permeable bags
no, but covered with lid
What is the incubation time and incubation temperature?
5 in, radiometric - 30 m
37 degrees centigrade, vary
ade, var dependent on as
y for broth; 21da
y for plates;at 37 degrees
o h it s 6-7 days
t ain used. For H37Rv plates are incubated 10 da
y , 37oC, incubation under hum
hu idified conditions. T
y recorded at the end o
y at 37°C for f or M. tuberculosis, 2 days
y at 35°C for M. smegm
ag plates and 2 w
te ts in 7H12 br
in 7H12 b oth and wi
oth and w th a macrophag
Which post-incubation reagents are added?
Additional comments
radiometric - chlorof
y eads on scanner
80, 1% v/v final concentration
Incubation time (hours) between reagent addition and reading.
N/A; spectro - kinetic m
at least 16 hours
t 16 hour usually 24 hour
ala ar blue assay
25 hours for alamar blue
2-4 hours f or M. smeg
g atis and 24 hour
and 24 ho s f or M. tuberculosis
variable depending on the m
le depending on the odel us
Is the reading done directly in the culture plate or tube or is
an aliquot removed for reading?
Additional comments on readout
500 cl aliquots rem
emission 590nm, excitation 530nm.
radio - quantification of react. product
15s for luminescence per well
scanned on scanner for visual density
If using a non-CFU readout, what is the signal:background ratio
of drug-free controls?
ee ontrol=0.45 to 0.47; Medium
ol=0.45 to 0.
Don't now, qualitat
ee D600: 0.20 –
: 0.04,Signal:Background = 5 -
Background ratio: 20
tio: 20 or M. smegm
Drug-free control
What variables, if any, have you found to affect this ratio?
e o growth and inc
ubation with alamar blue reagent.
Spectro - purity of
ate , source (vendor
e ipitation of the com
pound might give fals
a e negative, inc
80 Conta ination Precipitation of com
tion is not clear
If doing fixed concentrations what concentration do you use
and what is your criteria for a "hit"?
30 uM Hit: spectro 30% inhibition r
adio 50% inhibition
vary dependent on tes
10-fold difference betw
0.1, 1, 10 less than 1 is
a s per ml giving
g 90% or greater inhibition
do not screen in T
30 ug/ l as the highes
de inition of a hit depends on
the oject,t usuall
SAR purposes. Acives in the range of
y ollowed up.
No. Serial dilution for all compounds
ollowing concentr
05 icroM using 2-fold dilution pr
< 0.12 g/l is
considered as a hit.
No such things as
a a "hit" conc
greater than or equal to 90
equal to 90 inhibition
What positive and negative control drugs do you use?
INH and DMSO alone
y in isoniazid etham
positive control dr
o wing drugs depedent
ampin, Isoniazid, Moxifloxacin, Cefatax
n/a (not screening unk
Positive INH and Rif Negative is DMSO
For anaerobic assays
onidaz le as positive and INH as negative
INH, and Fluoroquinolone. Negative control simply s
INH, RIF, Moxi, PA-824 etc. Usually depends
INH as positive control
p in and Isoniazid
p in, levofloxacin, amikacin and linezol
y in and vancomyc
y in are used as negative contr
Drug-free culture control Media c
Variable depending on the c
able depending on the lass of
the ug under investigation
Positive: RMP, INH, SM, Mox, PA-824 Negative: metronida
What do you consider to be acceptable values for your controls?*
y in: 0.1-0.4 ug/m
0.4 ug/ l isoniazid: 0.025-
etha butol: 0.75-1.5 ug/m
plus or minus one dilution s
ted MIC values of each c
Positive control = 10-
Don't understand qu
MIC values stated b
INH 0.04, RIF 0.02-0.03, Moxifloxacin 0.1, PA-824 0.5
INH MIC 0.03-0.06 m
Rifampicin: MIC50 of 0.
10 concentration tested/as
ay rom 0.1uM to 0.00
INH: MIC50 of 0.25µM (
10 concentration
o 2.5uM to 0.004uM)
y CLSI are considered to be ac
Inh MIC 0.03 /- 1 tube Rif MIC 0.03 /- 1 tube
published values from
o Collins and Franzblau, 1997 an
*Note: this question was not clear to all respondents
Please indicate how you calculate MICs.
visual read MIC 90
tw old dilution tests
concentration of dr
t esults in luminescence lower than a 1:100 dilution of
100-fold difference
Broth MIC will be based on the w
the w l with no reduction of alamar blue dye.Agar
MIC will be based on dis
containg 7H11 plates.W
tw e is use to calculate MIC50/90
ion of growth.
Concentration inhibiting growth b
, also look at IC50 and s
ope o curve.
2- old dilution effec
on entration to inhibit growth c
Low t concentration to prev
ation o visible sediment at bottom
o graph plotted w
aph plotted w th PRISM, compound ser
ial diluted (10 dilution needed to der
on needed to d ive a proper curve)
aph, w th sigmoidal curve is
plotted to estimate the M
Y axis will have the OD600 values and the X-
axis will have log
The MIC is the lowes
t oncentration that inibits 99% of the bac
teria present in the inoculum.
interpolation using in-house s
inhibition relati
ve to drug-free c
Do you conduct screens at fixed concentrations of a drug or do you
determine MICs for every sample?
MICs for every sample
Fixed concentration screen, many drugs in a preliminary screen. MICs for anything promising.
determine MICs for every sample
Usually we do MICs
I don't understand the question
MIC for all the samples are determined
MIC for every sample
limited samples involved, do MICs
Always do MICs.
MICs for every sample
fixed for the inital screen and then determin "exact" MIC for hits
Fixed concentration used for medium/high throughput cell-based screening. Done against BCG at 10 M.
MIC is done for every sample.
Usually large screens start with fixed conc. at 16-32 ug/ml. After that the positives are tested for MIC
MIC for each sample
Usually depends upon the number of compounds in a series or library. For large collections, run primary screen at fixed concentration. For smaller sets or high predicted activity, do MIC for each sample.
What variables, if any, have you found to affect the MIC for standard
anti-TB drugs or experimental compounds using fixed concentrations?
Incubation time and inoc
y an be an issue for
to be onsistent for
pin' MIC against H37Ra is 10-fold lower
old low than against H37Rv.
these are very good dr
onsistent MICs. Media age may b
at had rashed out a
the ells, initial OD.
There is a tendenc
a tende y to pick
e po itives since the te
t in singlicate, wh
ate, w ich could be due to
Unreasonable question
poun ; serial dilution and m
on and ixing.
Regarding high throughput screening:
which types of compounds do you use?
What is the largest library that you
have screened or plan to screen with
your assay?
What is the source of your HTS library and in what format
is the assay done in (e.g. plate, vials, powder, etc.)
HTS library and in
at the assay done in (
e.g. plate, vials, pow
o mercial library; s
he ical, natural products, ex
ua y done in plates
Chembridge, Analyt
y icon, Broad Institute a
itute a DMSO stock
All in microtiter plates
t: ripos and Nanos
Novartis archive, c
d available in liquid (plate)
are in plates and ar
AZ corporate collection. 38
at u ing RBMA. Compound s
n't have any library
ChemBridge NovaCore, Prestwick, ECUM ac
tino extracts, all in 96 wel
What is the type and make of tubes, plastics and glassware
used for the stocks?
(Matrix) or glass vials.
supplied by vendor
Still to be determined. Most lik
Glass vials of 3m
96 w ll flat bottom U shape plates
Appropriate kind of
Additional responses
and intraplate controls
Please give examples of any methods done to normalize your procedures.
For instance, are there control samples included in each plate or do you
adjust for a certain level of background? Please explain.
Percentage inhibition calc
a ulated against no dr
e cence data not c
percentage inhibit
ion can be. Known dr
ugs included in each assay (separate plate) and their MICs used to indicate that as
ugs are included and f
h plates, negative and
y be included. Assays
ay will be done at several concentrations. DMSO
will be at least two
t tw DMSO plates with DMSO in all
INH totration on each plate f
h plate or "k
" nown" MI
C value on standar
U/ml final assay c
p in) and negative c
o solvent alone) in each plate.
igns are done with the control plates depending on how
ed. F large series, we us
aregular frequenc
, if screenign is only 300 c
o pds or so the tes
See our publications
Positive and negative controls are included in each plate. Standard drug c
a e included in each experiment.
What is your assessment of potency? For example is your assessment of
potency based on percent inhibition, percent activity, or other?
Do you include controls without drugs or without bacteria?
Percentage inhibition based on control without
teria controls also included.
y ontrols without drugs
Percent inhibition in terms of
without drug and without bac
ug and without ba ter
Percent inhibition and both c
on and both ontrols
y oncentration required to inhib
e. All plates have posi
o (with drugs, either rifam
p in or isoniazid)
z and negarive control (
o bacteria alone without the drugs).
y is based on per
cent inhibition. We inc
w thout drugs and c
w thout bacteria.
Percent inhibition. Media control and untreated gr
QC without drugs. Potency
y is expressd in MIC, MBC, MIC/MBC ratio, etc
percent inhibition Controls include: standar
w th bacteria standar
t ia bacteria only
How do you evaluate your assay performance?
Reproducibility of
Do you determine MBCs by sub-culture from the MIC test or
do you conduct as a separate test?
t 3 questions refer
u bactericidal concentrations (MBCs). Do yo
u deter ine MBCs by s
y ub-culture from
o the MIC test or
deter ine MBCs from
do not routinely per
MICs are determined in f
ned in ollow up s
y rom the MIC plate s
MBCs are done by s
y ub-culture.
If you run the MBC assay as a separate test, please indicate any variables
(e.g. CFU, luminescence, fluorescence etc) that differ significantly from
the MIC result.
done on 1 L cultures at
y ilution plating on 4" quad plates
w th 7H11-OADC agar
oth to agar.
Same setup for MBC determination
MBC plate will not receive the dye. G
oes f or CFU enumeration on
How ver, an addition
al plating is also don
What is your criteria for interpreting the MBC result
(e.g. percent killing of the starting inoculum)?
o include a control wi
w th no added drug to ens
ill of starting innoculum
ent illing of the s
counting colonies to determine CF
U/ml for given di
t ting inoculum, MBC90 as
ent ation to achieve 10-fold k
old illing and MBC99 as the concentration to achieve 100-
Interpretation of
etation o the MBC result is based on per
t ting inoculum.
MBC is the minimum
t ting inoculum.
90% reduction in cfu relative to the starting inculum
Please describe in as much detail as possible your
M. tuberculosis strain choice for non or slowly replicating (NR/SR)
assays and inoculum preparation.
MTB H37Rv exponentially
y growing culture, grown in r
oller bottle, OD600 of
600 o 0.2 – 0.3
and additional 0.03% Tween
edia to inal OD600 of
600 o 0.02, aliquoted to tubes
ubated at 37oC, 130
ay etup Nutrient star
t vation setup: grown in 7H9 m
S and then cells pelleted by c
y entrifugation and
in Dulbecco's PBS containing 0.025% Tween
a inal OD600 of 0.1 and
0.1 and aintained in roller bottle at 37oC f
bottle at 37oC or 14 day
y obacterium tuberculosis, M
y obacterium smegm
y obacterium bovis BCG are usualy used for thes
Nitric Oxide and the Hypox
ia models are used to
to easure the activity of
new ompounds agains
low replicating TB bac
Use H37Rv harboring a luxAB
A gene on a shuttle vecotr. Inoculum
is prepared in a s
ed in a ealed ferm
ealed fer entor in 7H12 m
in 7H12 edium in whic
u ption is monitor
ed. Cultures are was
ior to use. Details in Cho et al, 2007.
Please your NR or SR assay: format, sample preparation,
diluent, real time or specific endpoint measures, dyes and reagents, time frame, controls, etc.
ethod in MTB protocols boo
w ll plates containing drug in
DMSO (MTZ = + control, INH = - control). Transfer to anaer
hen deter ine CFU by p
U by lating on 7H11.OADC
Glass vials Methyl
y ene blue dye End point
u in triplicates in flat bottom
6 w l plates. Sam
. Sa ple preparation: 14 day ol
cells Diluent: DMSO
Endpoint measurem
e ent: CFU plating Time f
a e: CFU will be meas
conditions at 37oC Controls: 100uM Metroinidazo
z le, 25uM Isoniazid
o itive and negative controls
respectively. Loebel
setup Format: 4 differ
ent concentrations
o drugs in triplicates in flat bottom
6 w l plates. Sapm
epa ation: 14 day
old Loebel cells Diluent: DMSO Endpoint measurem
U will be measured on
incubation at 37oC Controls: 100uM Metroinidazo
z le, 25uM Isoniazid
z are used as negative c
Drastic oxygen deplet
t ial cultures wa
lacing loosely c
y apped tubes containing diff
drug concentrations inside an anaerobic jar (BBL) along with anaer
obic gas generation envelopes
(using palladium catalys
y low-speed centrifugation,
w hed twice with 7H9 medium to rem
and resuspended in drug-free
w e determined by p
ned by lating on 7H10 agar to
evaluate the bactericidal activity. In the
odel, gradual O2 depletion induces dorm
y obacterial cultures. Briefly, af
adual O2-depletion, c
ultur without exposure to oxygen w
gen w e treated further
e e counted on 7H10 plates
y incubating cultures with 150 μ
donor – diethlenetriamine/nitric oxide (DETA/NO) – for 2 hours
2 hour , f ollowed b
at: 96 w l plate, sample pr
epration and diluent - sam
ating cultures.
What strain of M. tuberculosis is used for
NR and SR assays and why was this strain chosen?
What is the source of your strain?
ain o M. tuberculosis is used f
ed or NR and SR assays
Mtuberculosis H37Rv, BCG
MTB H37Rv from AT
A CC (#27294 ).Chos
equen e is available
y obacterium tuberculosis H37Rv obtained from
A CC. A resistant
H37Rv lux AB constructed in hous
A CC 27294 and us
y luciferase and ac
What are your positive and negative controls for the NR and SR assays?
positive and negative c
ontrols for the NR and SR as
onidaz le and isoniazi
z d for anaerobic as
& discolouration of MB
Controls include: INH, Metronidazole,
Moxi, RIF - all samples are run in duplicate.
p in, isoniazid, m
onidaz le.Nutrient starvation assay:
Positive controls
idaz le and negative control
o is isoniazid.
y drug and media only,
What is your estimate of your laboratory throughput for NR and SR assays per day?
y ur laboratory thr
oughput or NR and SR as
ough put assay: 5-
2 to 3 experiments
What do you feel are the major variables that affect the outcome of the
NR or SR assay?
Head space ratio and Abr
t ring and rpm, Pr
Variation is seen at times wi
w ng slight activity)
with experimental cmpd
Starting inoculum, steer
ing peed and anaer
y , source of the PBS, s
t ting inoculum and c
lumping (for nutr
starvation model).
ation o the inoculum
What do you consider to be the strengths and weaknesses of the
NR or SR assay?
is heterogeneous
Strength Good indic
ant My obacteria Weak
ea ness Difficult for HT
y is less reproducible than regular MIC assay,
are run in duplic
ength ayne: Identif
pound active against anaerobic
obi non-replicating persistent bac
that ould potential
z on is critical for
del to obtain eaningf
iability in the data. T
n iate between ac
linical sterilizin
z g activity, good thr
low obustness, har
Which animal species do you use or have you used for TB drug testing?
What is the source/supplier of your strain of animal?
the ource/supplier of your
on Lab or Charles River Labs
other investigator
s (collaborators)
Centr , Singapor
B eeding Center, France.
National Institute of
Nutrition, India
Harlan Sprague Dawley
Please elaborate on the advantages of the infection model chosen.
u eptible species wh
or ation and allow
tion and allow stud
dissemination and he
eseeding o the lung allo
w the evaluaiton of
Cheap than oth animal, easy to c
variable because of
y reproducibility
e aerosol is the m
e lective of the h
pathology and dis
he ed growth of the
w rapid drug screening.
BALB/c mice develop pr
table plateau in CFU counts (as op
lining counts obs
h h are cheaper.
v lable and to handle.
ause it's an outbr
. Each ouse is different f
Mice: easy to handle. les
s compound requir
ptl groups in parallel. B6 colony no
Please explain the disadvantages of your chosen animal model?
the selves and ABSL-3 containm
an b. C57BL/6 is m
ore sensitive to some drugs (tox
as all experimental m
ental odels, it is
an experimental m
o trate all lesion types
ug etabolism in rodents
are different and c
o s species bridging studies
/calculations have to be perf
have to be per orm
More variability
y in CFU counts in Swiss mice.
s questionable. Patholog
y is not similar to humans
ence at low level of microorga
e the variability in
esults since the mice are diff
o pound, it's needed to over
y the alternate phys
nate phy iological states presum
y experienced by
ed by Mtb in human host. Can onl
against intracellular myc
y obacteria. The
any the in vivo models is the lack o
PK studies are difficult due to lo
What route of infection have you used (intravenous, aerosol,
intramuscular, intraperitoneal, other)? Please elaborate on past
methods used and current methods and explain why you use the particular method
u guinea pigs with a very
vi ulent M tuberculosis in a specially des
igned aerosol exposure (Madison)
au e this route and dose o
and dose o infection allo
ws the infection and disea
a e to develop in a m
y imilar to clinical TB.
aerosol challenge: aerosol infec
natural route of
terial number is high, i.e. 2 week
IV, aerosol(because it is easier to perform
o convenience and reproduc
u e intranasal route of
e. his partially m
y imics the natur
ety onsiderations, intravenous route is not used an
o k with the intravenous route of inf
that the intravenous route
w to extrapolate the res
a tivities of antibiot
o mostly and in both s
pecies). Intravenous
aerosol inhalation using Glas-Col sys
Do you have a placebo diluent/carrier injection or
gavage control in your experiment?
Have you ever seen an effect of the diluent placebo
on the outcome of the experiment? If so, please explain.
placebo on the outc
p iment? IfI so, please ex
No, this has been tes
e w se with placebo gavage
No, inactive compounds
How is the strain grown (e.g. pellicle, passaged through mice, other)?
w the strain grown (
e.g. pellicle, passaged throug
n to id-log phase in liquid culture, homogenized, s
onicated briefly, and f
obtain a single-cell s
u pension before f
eezing at inus 80C and determ
nus 80C and deter ining the cfu
c on a thawed aliquot,
filtered bacteria through 5 u
Normal in vitro culture of H37Rv
pellicle and frozen as
he eed stock is then upsc
t es, in Proskauer beck
t ough mice, preserved as seedlots
liquid culture to late log, then passaged
t , frozen in PBS-
80, occaisionally passaged in mice
Is there a record of passage number kept for your M. tuberculosis strain used?
If so explain this information and detail the maximum number of passages?
Have you ever noticed a difference in results of a treatment study depending
on the M. tuberculosis strain used?
What media is used to propagate the bacterial culture and
what (if any) concentration of Tween is used?
oth w th ADCC enrichment
y erol/0.05% Tween
H37Rv is propagated in 7H9-ADS medium containing 0.05%
7H9 broth with 0.02% Tween
y erol, casitone and O
Is the M. tuberculosis culture grown every time prior to the infection? If yes, on what parameter is the inoculum based
(e.g. OD, Klett, etc)? Or do you work with standardized, frozen working stocks?
Please explain the rationale for the method used.
is prepared and s
t ed in small aliquots at m
diluted to the appropriate concentration bas
ation ba ed upon th
w a precise duplication of the
level each time an
an periment is perform
h e is no detectable c
hange in infe tiv
No. OD is not reliable because if ther
tock, culture is grown f
c 7H9 broth OADC T
in a 500 l flask only e
s and large aliquots are made o
e the D 600 approac
he late log (0.6-0.9). The titer
n irmed by plate c
ounts and the vir
ulence is checked in GKO
aliquots are then f
e then rozen at -
80 C and stored s
ed o that each ex
have a new liquot of k
and virulence. W
in our last batch, that the vi
ter prolonged stor
w th standardized
w king stocks. Mtb is grown to an O
0. 0.5 in 7H9-ADS medium, c
w hed once in war
e in w m 7H9 medium
e w hed pellet is resuspended
00 o 1.0 in 7H9-ADS medium
ented w th 15% glycerol and f
ol and rozen at -
800C. The viable count of
ultures is determined by p
ned by lating prior to
is diluted and sonicate
bath sonicator) bef
and elect the inoc
according to McFarland suspens
U aintained. Thawed, diluted and us
eat ent is not initiated unti
t infection, the phys
y iological status
Do you perform pharmacokinetic/pharmacodynamic assays before or concurrent with
your animal studies? Please elaborate on the methods, computational analysis, mathematical
model used and results obtained. Do you have methodology and data on bioequivalence to human
infection and would you be willing to share these results?
ing the e fficiacy the a com
o pound in vivo, we
or ed at the highes
ed at the highe t poss
ible dose to see eff
tablish appropriate dose (
e equivalent dose com
ugs) . Methods are f
v using 6-8 timepoints following single doses or stea
y state dosing, obtaining serum o
y ardiac puncture us
nNonlin. e have data on
bioequivalence for
1 t-line drugs and s
o bination drug studies
to rule out significant PK
on in vivo PK data in the rel
cy is tested, the
ed, the inNonLin sof
tw e is used to m
ed to odel the PK pr
ile and imulate a
ing egimens.PK/PD indices (AUC/
MIC,Cmax/MIC,T>MIC ) are then deter
ned at ea h dose in or
e pected to be e
a ious in the infection
y before or concurrent with anim
n onLin is used.
k oral bioavailability s
a e blood samples fro
o retro-orbital sinus at 15, 45 a
e either LC-MS or TB bioas
ay using a luciferase reporter strain) to estimate s
oncentrations.
Do you perform toxicity assays before or concurrent with your animal studies?
Please elaborate on the methods used and results obtained.
l and pathology of
y tissue (live, spleen,
Y . Dose at higher than antic
ipated conc for a wee
tablish dose and i
limited to behavior changes and gross necrops
y imilar to that described by CS
Prior to in vivo PK and efficac
y tudies, in vitro toxicity
y and in vivo tolerab
y tudies in the relevant anim
t icity assay agains
ent ell lines namely,
y cell line, suspension),
y ian Golden hamster k
y ells, adherent), HepG2 (Hum
u an Liver Carcinoma, adher
lioma (Rat brain glioma,
. Cell lines obtained f
A CC) -genotoxicity (Ames test)
te -cardiotoxicity (hERG binding and patch clam
y Maximal tolerable dose in mice. Doses are s
ed ba ed on the in vitr
y before doing anim
y in vivo per se are not c
Cellular toxicity f
y or VERO and J774 c
774 ells and in som
o e case HepG2 cells. Toler
ing oral gavage and visual observation.
Do you routinely test the ADME (absorption, distribution, metabolism and elimination)
of compounds?
al bioavailability t
orbtion, and test dr
ug etabolism.
Both in vitro and in vivo ADME assays
ed p ior to efficacy tes
inhibition using the VIVD fluorscent kit. Just be
o e stability and to
extent, protein binding and Cac
Regarding ADME, what assays do you use? What sequence of tests is
utilized and when in the pre-clinical sequence are these implemented?
o oral bioavailability
ing M. tuberculos
e ore any in vivo
efficiacy studies are initiated. For
IN VITRO: -solubility at pH
1.0 and pH 6.8 -PAMPA (ar
b ane permeability)
P 50 inhibition IN VIVO -single dose phar
o lowing intravenous and oral administration to
deter ine oral bioavailability and PK par
e,C ax,AUC,volume of
t ibution). All these tes
u ing the lead optimizat
iz ion phase.At pre-clinical stage, m
ten ive ADME studies
are conducted in r
luding tissue distr
t ibution studies
with radio-labelled compounds
y in vivo, we do PK (Cmax
a , Cmin, AUC, T1/2, T
o al clearance, In vi
vo: Balb/c mice - uninfec
ted and in ected m
e or DM and PK.
Beginning to use prior to any
y ouse testing: 1)
4 inhi ion 2) human and
Caco-2 passage and
At what stage in the process of drug development is formulation optimized and finalized?
MIC determinations and c
o mulation is optimized at
lead declaration,
and finalized dur
e-clinical phase.
During the pre-clinical phase for in vivo use.
Late lead optimisation af
hortlisting a few
Do you routinely test colonies for drug resistance?
What inoculum do you use for infection of each animal model used in your lab?
u e an inhaled and ret
200-300 cfu delivered to mice.
in general, our aim is to impl
p ant 5x10e3 CFU in the lungs and begin tr
e aerosol (50-100) CF
U implantation at day 1 af
U depending on the pur
7 log10 CFUs.
osol: 10 cfu/anim
ol: 10 cfu/ani al G
M. tuberculosis Erdman
Do you consider your model to be a low or high infection dose animal model?
not high, not low
e tion dose animal m
e; Parenteral: high dose
What is the rationale behind the inoculum chosen for your infection model?
The normal infectious dose for humans is likely to be very low, since a single, primary,pulmonary lesion is almost always observed.
I think about 10 cfu is ideal. but 200-300 cfu/mouse at inital aerosol challenge is good to test Drug candidate because the bacteria grow enough number in the lung and spleen.
More like human in that disease becomes chronic
to obtain a bacillary population close to the bacillary population in human TB
most closely mimics natural infection.
high dose of 3.5-4 logs is implanted in order to have a burden of 7-8 logs at start of combination therapy 14 days later. in this way, it takes approximately 6 months of treatment with the standard RHZ-based regimen to cure all mice. low-dose aerosol may be used for monotherapy trials or LTBI models to prevent emergence of resistance or to better represent the lower bacterial burden of LTBI.
not sure, done for us by collaborator
As we have an intranasal, low dose infection model, the chosen inoculum is the lowest which can establish a lung infection in our animal models.But if very low inoculum is used , chances are there some bacteria get trapped in the airway and not reach the lungs (standard deviation was more when less than 5 X 102 bacteria was used.
We want to achieve a large bacterial population such as in human cavities at the time we start treatment.
In the case of aerosol infection model, low dose challenge is sufficient to establish the infection and is presumably close to natural conditions fo infection in humans (which is thought to be as low as 1-3 cfu). human mimic
How is the infection inoculum for every infection experiment checked for accurate bacterial numbers?
Do you plate the inoculum suspension? Do you enumerate bacterial numbers early after infection
(such as the day after infection) and prior to treatment? Tissue from how many animals/organs are plated?
The bacterial numbers are known from the cfu determination on the frozen inoculum; the reproducibility of this method over decades has been remarkable; there is no need to perform Day 1 lung cfu determinations unless the challenge strain changes
We validate each bacterial stock batch. - cfu numbers after aerosol challenge. - 3 mice, two times. There was almost no difference of
bacterial numebrs among same batch because we removed the clumps by filteration.
Plate lungs at t=0
We do everything; CFU count of (i) the suspension to inoculate (ii) lung the day after infection and (iii) day of treatment initiation
We plate the inoculum in serial 1:10 dilutions. We do a day 1 enumeration of the lungs and a pretreatment enumeration (typically 21 days after infection) of 5 mice.
we plate inoculum suspension and enumerate bacterial CFU the day after infection as well as the day of treatment initiation. we typically
culture the organs from 5 animals.
not sure, done for us by collaborator
We plate the inoculum suspension for every infection experiment.The bacterial numbers are enumerated after each infection like the day after the infection as well as on the day of treatment initiation. 5 mice each will be sacrificed one day after the infection and prior to treatment.
We plate the inoculum suspension. We enumerate the bacterial numbers one day after infection and on the day we start treatment in 5 to 6 animal organs.
Seedlot system for which the cell number is known and is nearly constant over a very long period of time at -70C. Additionally, before and
after infection, the inoculum is plated to ascertain consistency. For most expts, first day sampling is not done. However, for mutant studies, day 1 sampling is done. In all drug treatment studies, pre-drug sampling is done. four animals per group. Only Lungs plates.
Inoculum suspension is plated on the day of infection. Five mice are sacrificed on day 3 post-infection and 7 mice at the start of treatment
of day 10 post-infection.
For testing of single compounds and short term experiments, do you plate only one lung? Please explain.
We usually plate the homogenate of the same large lobe (usually lower right)from each animal; if very low numbers of bacteria are expected, a larger portion (including the whole lung) can be homogenized and a sizeable fraction of the homogenate (one-third to one-half) plated.
Both Lung and spleen.
4 mice per time point per concentration
We may use in that case more limited inoculum. and plate only one lung when the other is used for example for histopathology
We plate serial dilution of the whole lung for testing compounds
No, we plate both lungs.
In most expts, both lungs are plated. If HP samples are needed, lungs from satellite animals used for PK are collected.
No, whole lung is always used.
When organs are fixed for pathology or other purposes, which type of fixative
do you use for the tissues? Please elaborate on the methods for fixation
including how long the tissues are fixed.
y are harvested and mainta
y oaking in 0.9% Sodium Chlor
r alin fixative.
Have you ever noticed lesion-specific pathologic or microbial effects in any of your drug treatment arms?
Yes, in the Guinea pig model we see remaining TB bacilli present in the necrotic region of the primary granuloma after extensive drug treatment (for instance with TMC 207)
Yes. Meteorism is found in mice treated with high dose of moxifloxacin.
Have you ever observed cavitation or necrosis of lung lesions?
Do you perform any pre-sensitization of the animal to induce cavitary disease?
No. Cavities do occur in guinea pigs, but infrequently
No even when tried to sensitize mice
The guinea pig shows necrosis in the primary granulomas and after infection with certain clinical strains shows cavitation and mineralisation (no liquefication). Some necrosis is seen only in later stages of infection in mice. We do not do presensitization
No. We have never observed cavitation or necrosis of lung lesions in mice. We do not perform any pre-sensitization of the animal to induce cavities.
Not in mice. Caseation in guinea pigs
Are there other outputs measured to confirm or supplement the CFU data?
Please check all that apply.
Regarding long term trials: Do you plate: 1) all or only one lung lobe?
2) total spleen? 3) other organ(s)? Is the entire homogenate plated?
We normally plate a fraction (one-tenth) of the homogenate from one large lung lobe and half of the spleen;
1) all lung lobe 2) total spleen
All of lung, all of spleen
Total lung and sometimes total spleen too
For short term trials usually with single compounds, we collect left lungs and spleens, transfer in 4.5 mls of saline and plate 1:5 (0-7). For long term trials looking at sterilization of compounds, we collect whole lung, transfer in 1 ml and plate completely. Same for spleen.
we homogenize both lungs in a total of 2.5 ml PBS and then plate the entire homogenate on 5 7H11 plates (0.5ml/plate)
we do not do this
The entire homogenate is not plated. Whole lungs and total spleen are asceptically removed and ground using a tissue homogenizer in 4 ml of Phosphate Buffered Saline (PBS) supplemented with 0.05% Triton X-100. We take 25ul of the homogenate , serial dilute (diluentused is 7H9+ADS medium) it and plate 50ul from each dilution (we plate 50ul of undiluted homogenate as well) This we extrapolate to entire homogenate (whole lung).
We plate 2 lobes and also the total spleen. The entire homogenate is plated if the treatment is potent and given for a long duration and we
expect few or no bacilli.
Total mice lung plated. Entire homogenate plated without dilution. Homogenised in 1 ml volume and plated on 5 plates per animal.
Entire lung is removed, homogenized in 3 ml HBSS and serially diluted in HBSS. One hundred microliters of undilute or diluted suspension is plated on 7H11 agar in duplicate.
In a long-term sterilizing experiment, what fraction of the organs is plated?
What is the volume of diluent used?
In a long-term sterilizing experiment, what fraction of the organs is plated? What is the volume of diluent used?
One-third to one-half; 5 ml (L);2 ml (S)
Lung and spleen, 5ml
Entire lung in 2.5ml of PBS (5 plates)
the whole organ and 1 ml diluent
all of lungs in 2.5 ml PBS
we do not do this
Assay not established in our institute.
The entire fraction is plated (2.5 ml ).
Total lung in 1 ml.
If not all the tissue is plated, is this mentioned in your methods of publication?
we do not do this
If the entire fraction is plated, this is mentionned in publications. If it's mentionned in the publications that dilutions are plated this means that not all the tissue is plated.
Have you ever done an experiment designed to test relapse of a regimen? If so, please elaborate on the time between
the end of treatment and final sacrifice of the animal. Please list any immunosuppression agents utilized. Can you
describe methods used to assess bacterial burden (e.g. CFU counts, gross observations, PCR etc)?
What statistical method was used?
Yes; up to 20 weeks following cessation of the drug regimen; with and without immunosuppression (hydrocortisone; cyclophosphamide); clinial signs, cfu, histopathoology
from 12 weeks to 40 weeks after the end of treatment. we check the bacterial number by CFU counting. We used Dexamethason. Mice
received subcutaneously Dx.
Of course, it is the basic method to assess cure: 3 to 6 months after treatment completion, culture of the entire lung (and/or spleen). Standard statistical method
I have done very few relapse studies up to date. The few studies involved 3 months of relapse time after cessation of Rx. In only one prior drug study (rifazalil) I used dexamethasone as immunosuppressant and check with PCR for remaining DNA.
yes, 3 months post-treatment, no immunosuppression, we plate only the undiluted homogenate and cannot quantify counts >1000 CFU.
We compare relapse proportions using Fishers exact test, controlling for multiple comparisons as necessary.
we do not do this
We have not done an experiment to test relapse of a regimen.
In the experiments we carried out to test relapse of a regimen, the duration between the end of treatment and the sacrifice of animals is always 3 months. We don't use immunosuppression agents. When measuring relapses, spleen are weighed, gross lung lesions are scored and CFU counts are determined by plating the entire homegenate of the lungs or a spleen on media. The relapse rates are compared using the Fisher exact test or the Khi2 test.
Yes. Post completion of exptl Rx, 4 weeks of 'sterile' period, followed by dexa 0.5 mg/animal uid i.p. for 4 weeks, fol owed by 2 & 4 week
sampling for relapse.
Have you ever obtained unexpected data using your models which contradict published
results or results presented by others? Please elaborate.
Metronidazole and PZA are drugs that have shown activity in some labs in mice but were not effective in our lab.
not really, although we typically observe antagonism of INH on the RIF-PZA combination and this has not been well explored in other models.
we do not do this
CFU counts are below limit of detection after only two weeks of treatment with isoniazid (10mg/kg) and moxifloxacin (100mg/kg)
If you have had unexpected outcomes of an animal trial 1) were you able to track down the problem?
and 2) how was it rectified?
we do not do this
We had unexpected outcomes in animal trial due to human error ( dilution, plating, labelling ) which was tracked down and rectified.
Have you any evidence to support or refute the ability of yours or any animal
model to mimic human disease and predict treatment outcomes?
Anyone who knows clinical TB and reads the guinea pig literature will recognize the obvious relevance of this model
The mouse pathology has no similarity at all with the human pathology. Thus our aim is not to mimic human disease but to mimic the response of M.tuberculosis to drugs used at equivalent doses
Mouse models are certainly the best animal models for early preciminal evaluation of experimental compounds. However, as the mouse model has its limitations (only one lesion type is present and all bacilli are intracellular) we feel there is a need for an additional animal
model to address these issues and to confirmation results.
it takes approximately 6 months to cure all mice with RHZ-based regimens. the sterilizing activity. the sterilizing activity of R and Z are very evident when they are removed.
we do not do this
The acute mouse model we use has severe limitations in reproducing human disease: high bacillary load during the chronic phase, lack of a true latent phase, structure of the granulomas, lack of caseation and fibrosis, overall course of the disease. Having all of these shortcomings in mind, we use the model as a means to confirm in vitro potency and PK of our compounds in an in vivo system, without making quantitative conclusions as to the magnitude of the treatment response. Despite its many flaws, we found that the mouse is a reasonable compromise to qualitatively and rapidly assess the efficacy of
new compounds in a medium throughput manner.
The only evidence that supports the ability of our animal model to predict human outcomes is the fact that with our model the RHZ regimen displays almost the same relapse rate as in patients. This relapse rate is never observed in the aerosol model for instance.
the biggest problem in tb drug development
Do you routinely test any control drugs in every experiment?
How is the reproducibility of these results from experiment to experiment?
Yes. We use INH, RIF, Moxi, Gati. We got similar data until now.
Of course we have always negative and positive controls, and are amazed of the reproducibilty of the results when all parameters are similar
In early testing of compounds in the GKO model, INH at 25 mg/kg is always included (published in GKO paper). For long term combination
treatment trials the standard regimen INH-RIF-PZA is included and this regimen (2 log killing in the first 3-4 weeks and 3 more logs in the next 4 weeks). All controls are reproducible in the mouse.
high reproducibility
Yes, we test control drugs (Rifampicin 10mg/kg or Isoniazid 10mg/kg) in every experiment. Rifampicin shows significant potency to control Mtb infection in this model. We observed 1.5 – 2 log10 bacterial load reduction in the lungs after 14 days of treatment and 2 to 3 log10 reduction after 28 days of treatment .The infection in the spleen completely controlled by Rifampicin , the average bacterial count being below the limit of detection at 2 and 4 weeks
post infection. For
Isoniazid , after 14 days of treatment the
bacterial load comes below the
detection (80 bacteria)
We test isoniazid in every experiment. The activity of isoniazid used in the same conditions (inoculum size, established or non-establised
infection, duration of treatment, frequency of administration) is quite reproducible.
Rif and Inh. Excellent reproducibility.
rifampin at 10 mg/kg and in one project, PA-824 at 100 mg/kg is used instead. Reproducibility is good with rifampin but can vary with PA-824 from clearly bactericidal to nearly bacteriostatic
Numerous anti-tuberculosis agents have been studied in the past decades. It is possible that there is information buried
in these important studies that would be useful to us today. Can you think of antimicrobial agents and/or models
that would be worthwhile revisting today? If so, please give your rationale for this and pertinent references.
Some earlier relapse studies used a low number of mice, which could/should be revisited using a large number of mice.
yes, including catabolic steroid antibiotics, plant extracts, urea classe compounds and spirodecane class compounds.
We have recently revived the rat model of tuberculosis infection, based on the following observations and results. Recent studies (Elwood et al., 2007; Sugawara et al., 2004b; Sugawara et al., 2004c) produced encouraging results in terms of histopathology and colony forming
units (CFU) profiles in the lungs of M. tuberculosis infected rats, that suggest the rat may indeed be a useful model for TB research in general and more specifically for testing new drug candidates. In addition, the tuberculin test is feasible in the rat (but not in the mouse), and a large array of immune reagents are available. Mtb infection in rat lung exhibits both an acute phase (20-25 days of exponential growth) and a subsequent chronic phase (stabilization of the bacterial counts). The chronic phase is induced, as is the case in the mouse, at the onset of the cell-mediated immunity and can last for an extended period of time (Orme, 2003; Sugawara et al., 2004b). Mtb can reactivate and cause a premature death of the infected animals when there is a failure of the immune pressure (Sugawara et al., 2006). Preliminary results from Bifani et al. (Unpublished observations) suggest that endotracheal inoculation of mycobacteria is a simple way to infect lung rat, very reproducible and prevent the spread of aerosols in BSL-3 environment. Because it is relatively inexpensive, small, easyto handle, and has reasonably and accepted predictive value in toxicology and pharmacokinetics, the rat has been a species of choice for early drug development in the phar
maceutical industry. By using the rat model, in vivo efficacy
data could be directly correlat
pharmacokinetic/pharmacodynamic drug parameters and toxicology studies. We hope that once optimized, the model will provide a
system which is time, cost and compound efficient, to substantially accelerate the discovery and early development of new anti-tuberculosis molecules. References: Elwood, R. L., Wilson, S., Blanco, J. C., Yim, K., Pletneva, L., Nikonenko, B., Samala, R., Joshi, S., Hemming, V. G., and Trucksis
, M. (2007). The American cotton rat: a novel model for pulmonary tuberculosis. Tuberculosis (Edinb) 87,
145-154. Orme, I. M. (2003). The mouse as a useful model of tuberculosis. Tuberculosis (Edinb) 83, 112-115. Sugawara, I., Udaga
and Yamada, H. (2004a). Rat neutrophils prevent the development of tuberculosis. Infect Immun 72, 1804-1806. Sugawara, I., Yamada, H., and Mizuno, S. (2004b). Pathological and immunological profiles of rat tuberculosis. Int J Exp Pathol 85, 125-134. Sugawara, I., Yamada, H., and Mizuno, S. (2004c). Pulmonary tuberculosis in spontaneously diabetic goto kakizaki rats. Tohoku J Exp Med 204, 135-145. Sugawara, I., Yamada, H., and Mizuno,
S. (2006). Nude rat (F344/N-rnu) tuberculosis. Cell Micr
obiol 8, 661-667.
Possibly, the Lurie Rabbits, that had resistant and susceptible phenotypes. Also, in the 1970s D. Smith and D. Mitchison, used clinical strains from India to infect guinea pigs, these clinical strains produced two different phenotypes of disease One chronic and one more progressive. Can we use these two as models of human latent and active disease perhaps?
What statistical methods are run in conjunction with your animal experiments?
Do you consult with a statistician?
Main treatment effects are analyzed first by ANOVA; only if the F statistic is significant is a valid between-means test applied to the individual treatment groups.
T-test, anova test. We consult somtimes with a statistician at our university.
Alas yes and it is very important
One way ANOVA, followed by stringent methods for pairwise comparison such as Tukey test, Dunnett's test. For relapse studies we use include T-test (cured versus relapsed mice)
we conduct our own one-way ANOVAs and Fishers Exact tests, controlling for multiple comparisons. we do not consult with a statistician.
we do not do this
A compound is active in this model if the CFUs are 10 times lower (1 log10 reduction) in the treated vs. untreated group. We don't consult statistician.
We use basic statistics. We use parametric tests such as student t-test when dealing with quantitative variables such as CFUs, spleen
weights when we have all the conditions to use them such as similar variances otherwise we use non parametric tests such as MannWhitney. For the binary (qualitative) variables such as relapses, deaths, we use the exact Fisher test or the Khi2. We don't consult with statistician except if it's needed.
One-way ANOVA - Dunnet's Multiple Comparison Tests TTests
Source: http://mrl.colostate.edu/files/2013/01/survey41808.pdf
Österreichisches Institut für Familienforschung Austrian Institute for Family Studies 34 – 2003 „STAHLHART – MÄNNER UND EREKTILE DYSFUNKTION" Olaf Kapella working papers have only received limited review ÖIF, Gonzagagasse 19/8, A-1010 Wien Tel. +43-1-535 14 54-0 Fax +43-1-535 14 55 url: http://www.oif.ac.at email: [email protected]
DENTAL ASPECTS OF ENDOCARDITIS PROPHYLAXIS : New recommendations from the British Cardiac Society Guidelines and Medical Practice Committee and Royal College of Physicians Clinical Effectiveness and Evaluation Unit. 2004 [18-04-03] Graham J Roberts David Ramsdale Consultant and Professor of Paediatric



