Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Doi:10.1016/j.bmc.2006.06.024

Bioorganic & Medicinal Chemistry 14 (2006) 7011–7022
Drug Guru: A computer software program for drug design
using medicinal chemistry rules
Kent D. Stewart,a,* Melisa Shirodaa and Craig A. Jamesb
aAbbott Laboratories, Global Pharmaceuticals Research and Development, Abbott Park, IL 60064, USA
bMoonview Consulting, LLC, San Diego, CA, USA
Received 27 April 2006; revised 6 June 2006; accepted 8 June 2006
Available online 25 July 2006
Abstract—Drug GuruTM (drug generation using rules) is a new web-based computer software program for medicinal chemists thatapplies a set of transformations, that is, rules, to an input structure. The transformations correspond to medicinal chemistry designrules-of-thumb taken from the historical lore of drug discovery programs. The output of the program is a list of target analogs thatcan be evaluated for possible future synthesis. A discussion of the features of the program is followed by an example of the softwareapplied to sildenafil (ViagraÒ) in generating ideas for target analogs for phosphodiesterase inhibition. Comparison with other com-puter-assisted drug design software is given.
Ó 2006 Elsevier Ltd. All rights reserved.
A rich tradition of analog design strategies has evolved
for creating new compounds within medicinal chemistry
research for biological evaluation. When similar physi-
cal properties between lead compound and analog are
desired, ‘bioisosteric' replacements are commonly em-
ployed. Where more structurally altered yet still compat-
ible differences between lead compound and analog are
desired, non-classical replacements are considered. This
latter situation occurs when a chemist desires structures
Figure 1. Two well-known examples of rule-of-thumb for designing
that are outside of the intellectual property of a compet-
analogs: (a) the carboxylate-to-tetrazole replacement, (b) the amide-to-
itor or when attempting to achieve more dramatic
retroamide switch. Other examples are listed in or described in
changes in potency or bioavailability. Collectively, these
replacements are known to experienced medicinal chem-ists as ‘rules-of-thumb' for drug design. Two examplesof well-known design rules-of-thumb, the carboxylate-
sent a useful starting place in a drug discovery effort,
particularly when other knowledge such as pharmaco-
illustrated in have historically been considered
phore models, 3D receptor structure, or structure–
to yield analogs of high interest in medicinal chemistry
activity relationship data is limited, low quality, or
programs. Hundreds of these structural transformations
have been reported in the literature and have potentialfor general applicability and acceptance as design
We have written a web-based computer application,
rulesWhile no single rule is ever guaranteed to
called ‘Drug Guthat contains the historical rules-
achieve the desired endpoint, the traditional rules repre-
of-thumb as lines of SMIRKS code.The name ofthe program is an acronym for drug generation usingrules. The program applies a library of rules to any inputstructure and then permits visual or computational eval-
Keywords: Computational chemistry; Drug design.
* Corresponding author. Tel.: +1 847 937 1205; fax: +1 847 937
uation of the output structures. To our knowledge, no
previously described or commercially available software
0968-0896/$ - see front matter Ó 2006 Elsevier Ltd. All rights reserved.
doi:10.1016/j.bmc.2006.06.024
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
accomplishes all of the tasks of Drug Guru, and we pro-
The ring-break transformation (rule #9 in finds
pose that it nicely complements other computational
its basis in the general strategy to take atoms covalently
approaches. In this article, we describe the basic features
connected within a ring and replace with atoms that are
of the software, illustrate its use in a retrospective study
intramolecularly H-bonded to form a ‘virtual' ring sys-
of sildenafil, and end with a comparison to other com-
tem. Drug Guru has several of these ring-break rules.
puter-aided drug design software.
In the example described here, one of the aryl rings ofa fused aromatic ring is replaced with a carbonyl andamine that project from the other aryl ring. This ring-
2. Materials and methods
break rule was recently illustrated by Novartis in theirscaffold morphing discovery of the anthranilic amide
2.1. Drug Guru rules
inhibitors of KDR kinase.This rule and example areillustrated in
The most important and novel aspect of Drug Guru isthe collection of rules. We found the SMIRKS transfor-
The NC-switch transformation, rule #10 in ,
mation protocol useful for encoding rules.SMIRKS is
finds its basis within the general ‘carbon-to-nitrogen'
a linear text string that represents a graph transforma-
replacement strategy used in discovery of new heterocy-
tion which, when applied, converts a representation of
cles as core pieces in drugs. The general notion is to take
an input structure (a SMILES code) into a new structure
every carbon in a structure and sequentially replace each
(a new SMILES code). Ten illustrative rules and their
with nitrogen. In the specific case of rule #10, two atoms
corresponding SMIRKS codes are given in
are ‘exchanged' in the situation where a vinyl amine ispart of the aromatic system, leading to an ‘NC-switch'
In practice, two general kinds of SMIRKS are required
rule. A good historical illustration of an application of
to cover the transformations encountered in most
this NC-switch rule may be found in the research pub-
medicinal chemistry programs: functional group trans-
lished by Abbott Laboratories that led to the pyridone
formations and molecular framework modifications. In
class of antibacterial agents typified by ABT-719.This
the version of Drug Guru described here, there are 133
rule and example are illustrated in
and 53 rules in each of these general classes, respectively,for a total of 186 rules. A list of rule categories is given
2.2. Structure input
in Fourteen functional groups were empiricallyselected as most frequently encountered in medicinal
Input of a chemical structure can be via corporate iden-
chemistry research programs. Rules corresponding to
tifier code, drawing program, or by coordinate file,
entries 1–7 of are typical functional group trans-
either individually or in batch. Typical input structures
formations and will be familiar to most experienced
include the current lead structure for a particular pro-
chemists. A text mnemonic is assigned to each rule to
ject, a competitor compound or a natural ligand. After
permit ready comprehension of the general nature of
entering the input structure, the user selects the run but-
the rule , column 1). An extensive literature sur-
ton with no additional user information needed. All gen-
vey of medicinal chemistry reports is currently underway
eral transformations are applied in this mode. An
to produce a more expanded and comprehensive listing
‘expert user' page is optionally accessed to allow some
of rules and their corresponding SMIRKS. In addition
additional features (described below) to be explored if
to the functional group transformation rules, a variety
desired. The input web page is illustrated in .
of molecular framework modification rules are also
Tautomerism of the input structure was found to be
encoded, including ring break/form, ring contraction/ex-
an important factor: different tautomers give different
pansion, and ring replacement rules (Rules for
output results. When tautomeric possibilities are found
entries 8–10 in are representative framework
by an automatic tautomer check within Drug Guru,
modifications. Homologation rules, such as transforma-
the user is queried to select the desired input
tions that extend ring substituents by an oxygen, sulfur,
carbon, or nitrogen atoms (such as entry 8, areexamples of rules that currently fall into a ‘miscella-
2.3. Evaluation of output
neous' category (Also listed in withinthe category of framework modifications are rules for
In Drug Guru, the primary method of evaluating the
altering molecular conformation: for example, the addi-
output structures is by visual inspection. Currently, we
tion of geminal methyl groups to flexible chains to ex-
have implemented 186 rules and empirically observe that
ploit the Thorpe–Ingold effecTwo framework
a typical medicinal chemistry request will result in 50–
modification rules from the ring-break rule, en-
150 output structures. Structures with higher structural
try 9; and the NC-switch rule, entry 10, illustrate more
complexity, that is, high number of functional groups
complex and less obvious structural transformations
or skeletal connections, yield more instances of rule
and are described in detail below. A public web-based
applicability, thus leading to a greater number of output
utility program is available (
structures. Drug Guru requires only seconds for genera-
that will allow readers to evaluate
tion of results. The visual evaluation and thoughtful
the SMIRKS supplied in Drug Guru is written
analysis of the results by the end-user medicinal chemists
in Perl programming language, and uses the DayPerl2
typically requires 10–30 min. With the number of output
interface to the Daylight SMILES, SMARTS, and
structures in the hundreds, we desired a database man-
Reaction Toolkits.
agement tool that would conveniently permit casual
Table 1. Illustrative listing of rules
Rule illustration
[C,c:1]-[OH] � [C,c:1]-[O]-C
2]-[CH2]-[C,c:2] � [C,c:1]-[S]-[C,c:2]
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
Table 2. Rule categories and number of rules
not surprising since Drug Guru was programmed with
Functional group transformations
rules derived from the traditional medicinal chemistry
knowledge base. Even at this level, the program pro-
vides value due to its comprehensiveness. Importantly,
in test situations, chemists have additionally indicated
that non-obvious structures are also included, thus mak-
ing the use of Drug Guru an even more fruitful
experienced medicinal chemists gain insight from Drug
Guru as a starting place for learning about analog de-
sign. The references and example structures provided
with each rule provide a starting place for learning prac-
tical medicinal chemistry (see Rule History description
Drug Guru will occasionally generate low quality, non-
Molecular framework modificationsRing break
drug-like structures (three contiguous heteroatoms as an
extreme example). Such output structures result from
extrapolating specific rules to general situations, not
all of which are relevant, or a practical failure to consid-
er every possible situation to which a SMIRKS code
may apply. We have opted not to discard the low quality
structures: (1) in practice, the number of low quality
Total = 186 rules.
structures is small, less than 5% of the output, and nota major nuisance. (2) The unorthodox application ofrules sometimes results in very novel output which mightlead to an ‘outside-the-box' idea. Our current strategy
with output lists of less than 200 entries is to let themedicinal chemists themselves judge ‘goodness/badness'
of the structures, and the default usage is to not apply
any structural or numerical ranking. Further refinement
of the rules and/or addition of an optional computation-al filtering step will reduce the number of low quality
structures. In cases of output lists with greater than
200 entries or where other scientific information is avail-able, some filtering and/or ranking of output is needed.
Ranking based on calculated physical property is an op-
tion which is further described below.
Since it is possible that different rules can result in iden-
tical output structures, duplicate entries are grouped in a
final step prior to display of all of the results to the user
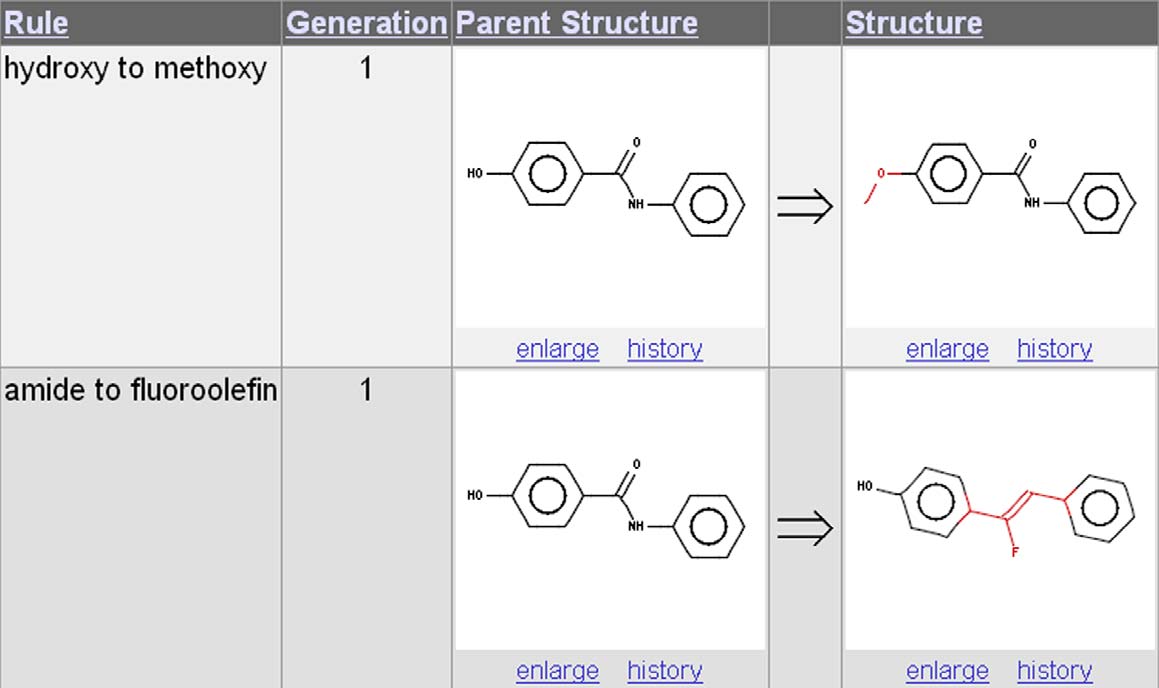
in web page format. A typical output page using tworules from is shown in . A ‘history' linkis available to provide information on the structural
transformation, for example, scientific basis and scopeof the transformation, examples of application within
medicinal chemistry history including proprietary cor-porate history, and any interesting unpublished lore
Figure 2. (a) Illustration of the ring-break transformation (rule #9 in
about the transformation. An example history page is
(b) Specific example of the ring-break rule in converting
shown in for the hydroxy-to-methoxy rule. In
PTK787 to the anthranilic amide analog AAL993.
addition to automatically archiving results in a user areaafter accessing Drug Guru, options for sharing results
inspection of this many hits and permit archiving for lat-
with other scientists or exporting data are provided.
er study. We selected the commercially available RENEdata analysis tool as one that (1) understands chemin-
2.4. Special features of Drug Guru
formatics and Daylight parameters, (2) permits searchand sort capabilities, and (3) uses Oracle-based informa-
While the default usage of Drug Guru by the medicinal
tion storage for data manipulation and archival.
chemist requires only input of a structure and pressingthe ‘Run' button, there are several user options that
Many of the chemical structures that Drug Guru creates
are provided on an ‘expert user' page. When an input
are reasonable target structures that, admittedly, will be
structure has more than one occurrence of the function-
obvious to most experienced medicinal chemists. This is
al group to which a rule applies, the question of how
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
Figure 3. (a) Illustration of the ‘NC-switch' rule (rule #10 in (b) Specific example of the ‘NC-switch' rule in converting the antibacterialdrug ciprofloxacin to
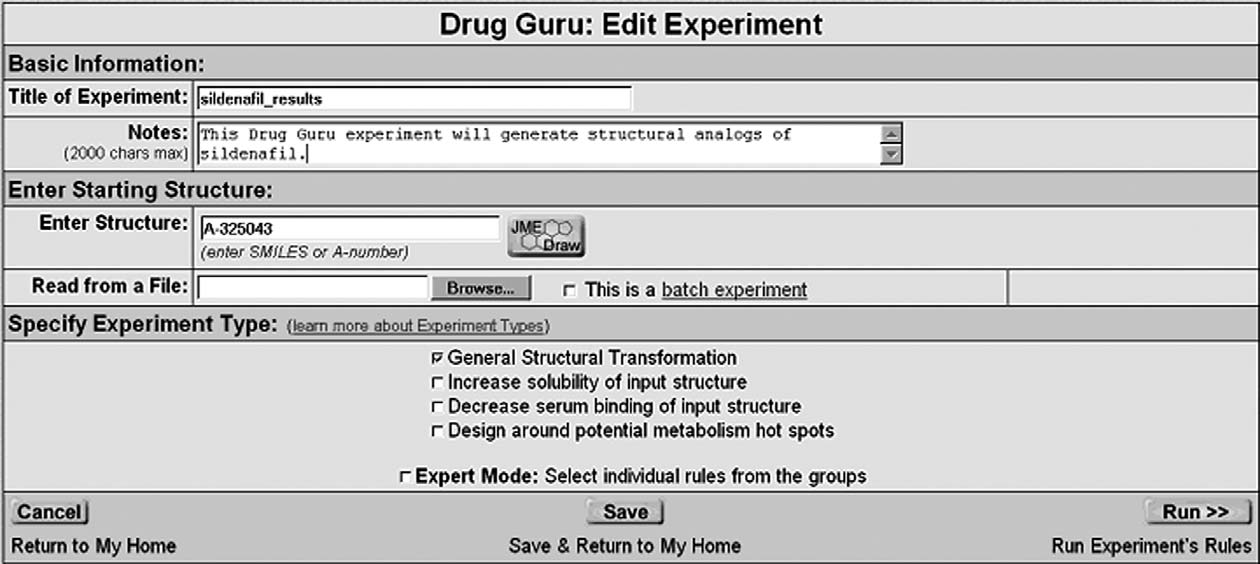
Figure 4. The structure entry web page of Drug Guru. In this example, the structure of sildenafil is accessed by entering the corporate code,A-325043, for this drug.
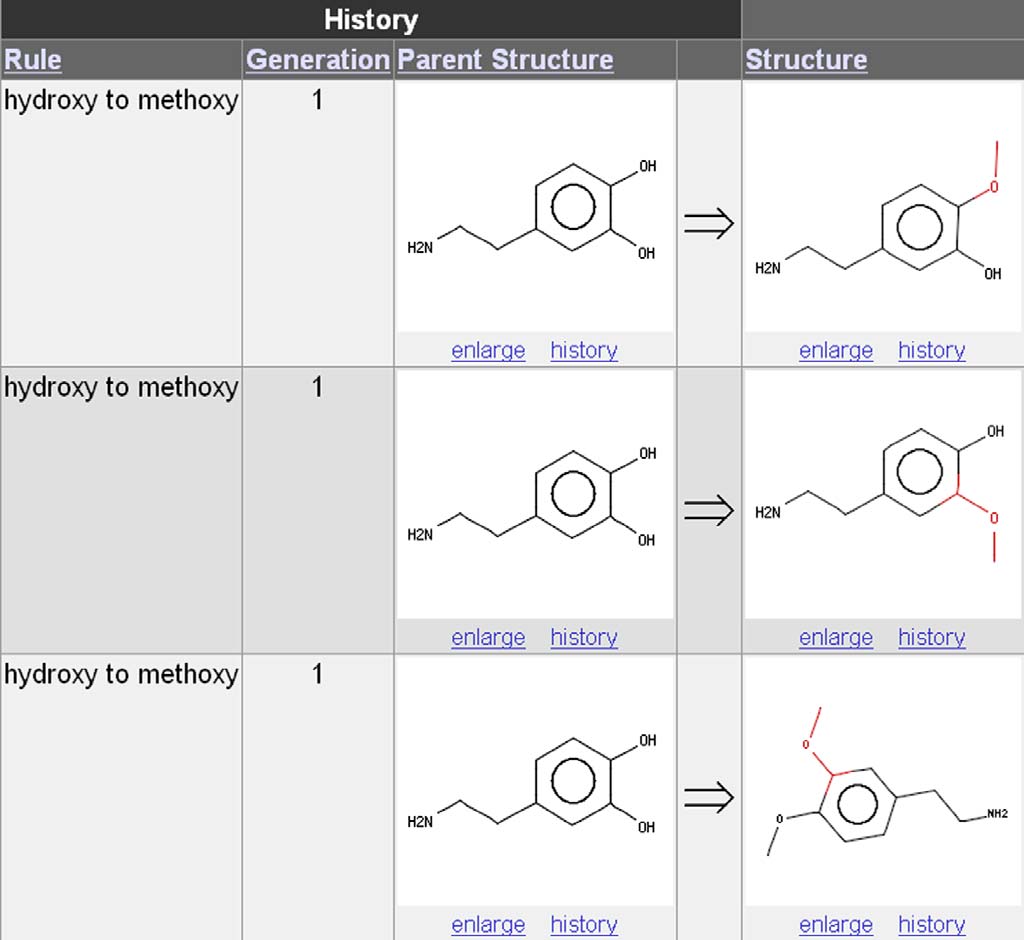
many times Drug Guru applies the rule needs to be an-
tion experiment is shown in In practice, the
swered. The example of carrying out the hydroxy-to-
combinatorial expansion leads to a very large number
methoxy transformation on the two hydroxy groups of
of structures (thousands) for a rule set selection of
dopamine, see best illustrates this feature. In
100–200 rules; therefore, the number of generations is
the default mode, only the two mono-methylated result-
currently limited to four.
ing structures are obtained. Optionally, an exhaustiveapplication of the rule produces a third di-methylated
Within Drug Guru, there is an option provided to study
structure. This differentiation between single and
the output list according to calculated physical property,
exhaustive application of a rule becomes particularly
such as log P, rule-of-5, polar surface area (PSA), or
important for rules involving replacement of hydrogen
rotatable bonds. In test studies with known drugs, no
atoms. In this case, selection of exhaustive replacements
obvious bias or uneven trend in the calculated physical
can lead to a very large number of output structures.
properties of the output molecules was evident: struc-tures with a continuum of both increased and decreased
Another user option is to allow multiple generations of
physical properties were observed with approximately
application of the set of Drug Guru rules to an input
equal frequency. A typical distribution is shown in
structure. In the default setting, a single round of trans-
using Gleevec as input. Sorting the output list
formations is applied. Optionally, the output structures
according to physical property is provided as a user op-
from the first round can be automatically re-submitted
tion and can facilitate identifying structures of high
for a second round of transformations. This has the
interest. Of particular value is the mathematical differ-
interesting effect of generating quite novel molecular
ence in property when comparing the input and output
structures for evaluation. An example of a two-genera-
structures. When the ‘D-property' column is selected
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
Figure 5. Example output page from Drug Guru using rules 2 and 3 from The output page contains four columns, with each separate outputstructure as a new row. Column 1 is the name of the rule, column 2 is the generation number, and columns 3 and 4 are the input and outputstructures, respectively, shown as a 2D depiction. Atoms directly involved in the transformation are highlighted in color to facilitate visual inspection.
This example shows only two output structures, but in practice, lists of 50–150 structures are common.
for sorting, the user can quickly tabulate all of the struc-
tion. In the application of 186 rules to the structure of
tural transformations that give a desired result. For
sildenafil, 91 output structures were created by Drug
example, users can see all of the changes that lead to
Guru. While discussion of the entire output list is be-
an increase in C log P or a decrease in PSA. In
yond the scope of this publication, a few illustrative
a D-PSA calculation is illustrated.
examples will be given.
As a critical aspect of Drug Guru, we note that the rule
Five of the 10 rules listed in are applicable to sil-
list is not static, and new rules can be added at any time.
denafil and yield output structures represented in
For example, new structural transformations reported in
. Five rules do not apply because their functional
the literature or proprietary research discoveries of poten-
groups are not present in the input structure. The
tial general applicability are excellent sources of new
homologation-C rule (extend every substituent on a ring
rules. There is also the capability of creating subsets of
by one methylene unit) gives several non-redundant out-
rules for special purposes. For example, in addition to
put structures and only two, 36 and 39, are illustrated.
the set of general transformation rules described above,
The ethano-to-S and ether-to-thioether rules give struc-
we have included separate sets of rules for increasing sol-
tures 62 and 69, respectively. In this example, the more
ubility, decreasing albumin binding, or decreasing meta-
interesting and diverse structures result from molecular
bolic liability of the input structure. These rules are less
framework changes. The NC-switch rule described
well documented and more anecdotal in character, but
above gives rise to two structures possessing new [6.5]
nonetheless, still very useful in practice. As an example
heterocyclic core rings, output structures 64 and 65.
of this kind of additional Drug Guru rule, one of the
Gratifyingly, structure 64 possesses the heterocycle
metabolism rules is a ‘pyridine-block' rule which adds a
found in vardenafil (LevitraÒ), a drug in the same phar-
methyl group to the ortho-position of a pyridine. This rule
maceutical class as sildenafil and shown in .
is based on the strategy to sterically block the facile oxida-
Another NC-switch output, structure 65, possesses a
tion of pyridines with an ortho substituent. An example
heterocycle not previously encompassed within sildenafil
from the Roche group of an application of this design rule
or vardenafil patents.In another example of a frame-
has recently been publis
work change, the ring-break rule results in output struc-tures 60 and 78 (only 78 is illustrated) in which thearomatic 5-membered ring of sildenafil is opened to give
an amino-ketone structure. An intramolecular hydrogenbond between the amino and carbonyl groups of 78 has
3.1. Example of Drug Guru applied to known drug system
the potential of forming a intramolecular H-bond, andthus mimicking the aromatic ring of sildenafil. As dis-
Sildenafil (ViagraÒ) is a phosphodiesterase-5 inhibitor
cussed above, this ‘virtual' ring strategy has precedent
approved in 1998 for treatment of male erectile dysfunc-
in other studies. No literature reports of studies of chem-
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
Figure 6. Example of Rule History Page.
ical systems related to structures 60, 65, and 78 in inhi-
ed drug design software: docking and scoring programs,
bition of phosphodiesterase-5 or utility in treatment of
such as DOCK, FlexX, AutoDock, GLIDE, Chem-
male erectile dysfunction could be found. Therefore,
Score, and GOLD, de novo ligand construction pro-
compounds based on these structures may represent
grams, such as LigBuilder, SkelGen, Ludi, GrowMol,
interesting targets for further analoging around sildena-
and SPROUT, scaffold-hopping programs such as
fil and vardenafil. The exact structure of vardenafil,
LeapFrog, EA-Inventor, and FEPOPS, and pharmaco-
shown in is produced from sildenafil by Drug
phore analysis programs, such as COMFA, Disco, Cat-
Guru in a two-generation run when both the homologa-
alyst, and GASP.However, these other programs have
tion-C and NC-switch rules are applied. It should be
operational strategies that are fundamentally different
emphasized that information from sildenafil structure–
from Drug Guru. Drug Guru does not rely on a pre-ex-
activity studies was not used in the creation of the rules
isting database of ligand structures or utilize energy-
of Drug Guru; therefore, these examples shown here are
based fitness functions to score or assemble ligands.
truly derived from the history of medicinal chemistry,
No prior structure–activity data are required. Drug
implemented as computer-coded rules-of-thumb for
Guru uses a set of pre-selected and well-defined medici-
nal chemistry rules to construct new structures. Rankingis optional and currently is based on calculated physical
3.2. Comparison with other computational chemistry
properties. ‘Comparison' of Drug Guru with the com-
puter programs listed above is useful mainly in thinkingabout synergy, rather than ranking performance capa-
The overall objective of Drug Guru is to facilitate drug
bilities. Many of these programs could conceivably be
discovery, a goal that is shared with other computer-aid-
used to help analyze the output of Drug Guru. We have
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
Figure 7. Example of an ‘exhaustive' application of a Drug Guru rule. The first two output structures result from a single application of the rule. Thelast structure results from application of the rule at all possible sites exhaustively.
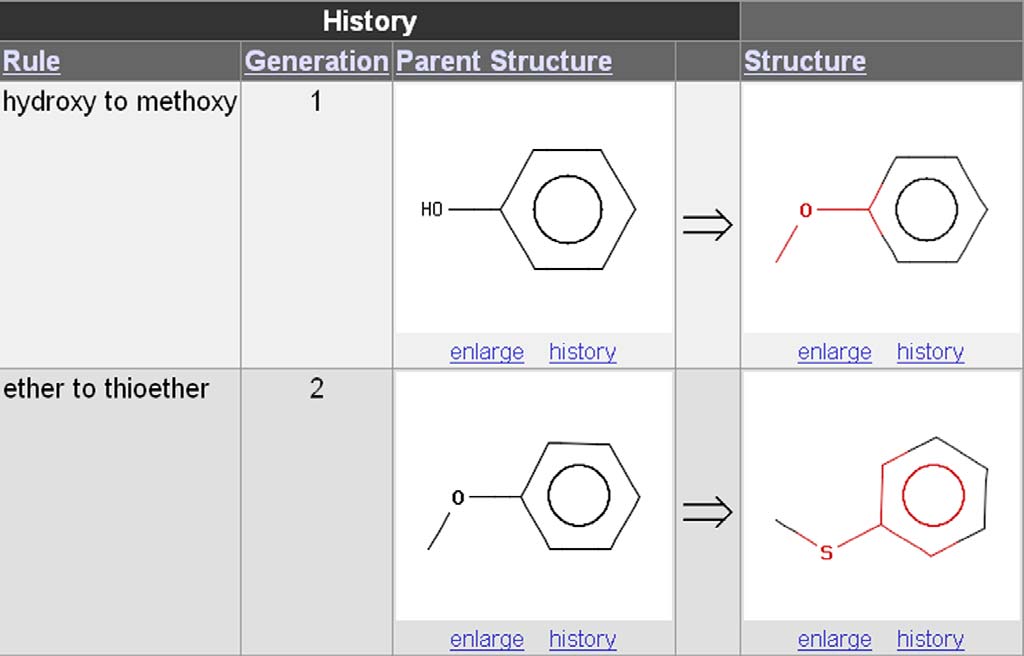
Figure 8. Example of a ‘multi-generation' use of Drug Guru. The generations are indicated in column 2. In generation 1, phenol is transformed toanisole by the hydroxy-to-methoxy rule. In generation 2, the anisole formed in generation 1 is transformed to thioanisole by the ether-to-thioetherrule.
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
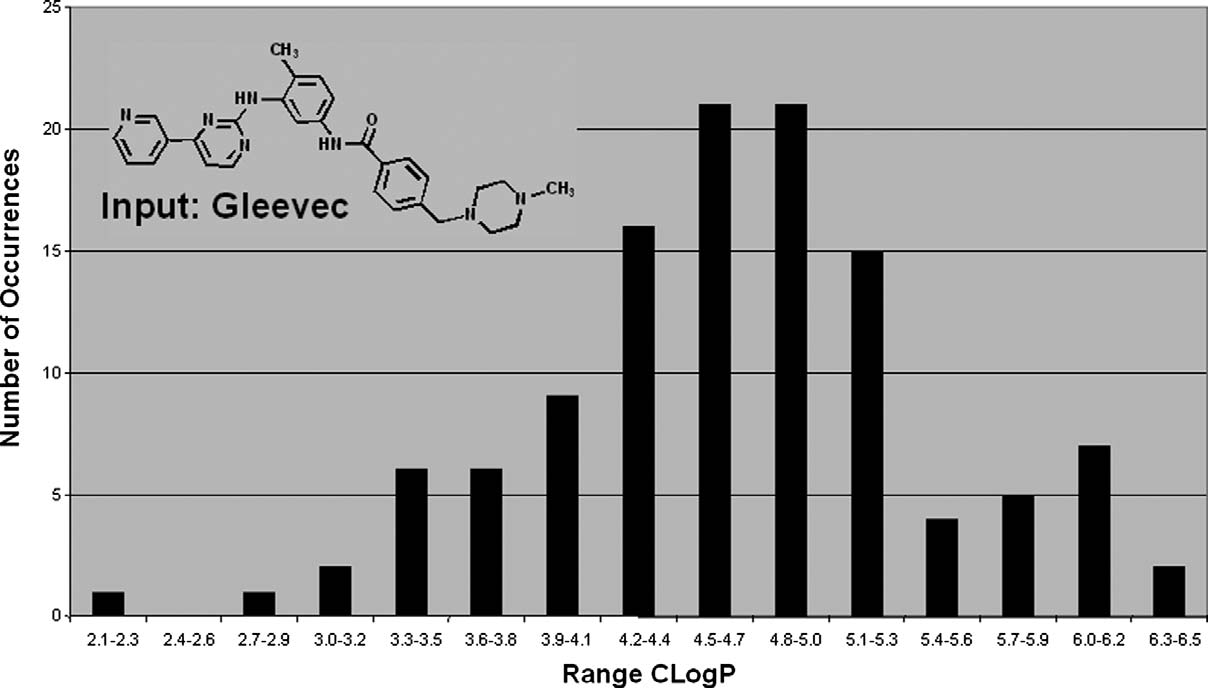
Figure 9. Distribution of the C log P values that are calculated for the 116 output structures resulting from using Gleevec as input structure. Gleevechas a C log P of 4.5 and would be located at the center of the distribution.
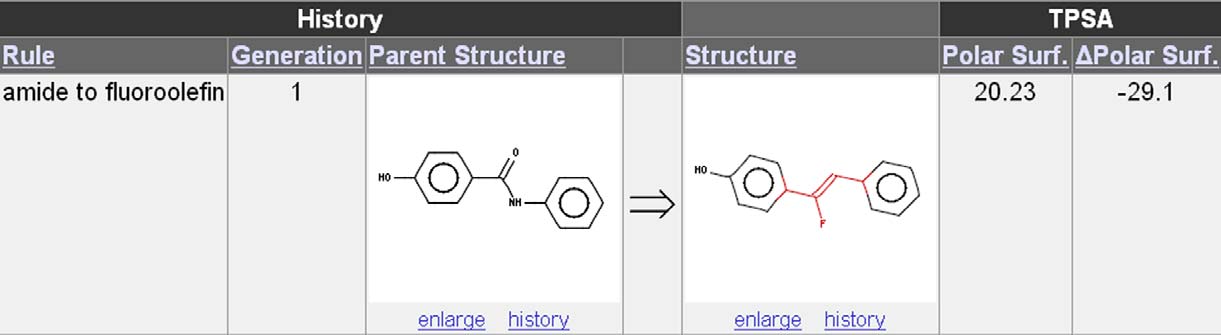
Figure 10. Example of calculation of physical property of a Drug Guru output structure. The polar surface areas, PSA, of the input and outputstructures are 49.33 and 20.23 A
˚ 2, respectively. The web page reports the PSA of the output along with the mathematical difference, the D-PSA value,
of 29.1. In this case, the decrease in PSA is indicated with a minus sign. This ‘D' column is available for all calculated properties and can be sorted tofind all structural changes that give a consistent change, for example, all transformations that lead to a decrease in polar surface area.
already experimented with taking Drug Guru output
in Drug Guru, BIOSTER compiles a large number of
directly into docking and pharmacophore programs
specific examples (14,300 bioanalogous pairs in the
for further evaluation (work not reported here).
2005.1 release). Drug Guru differs from BIOSTER inshowing the structural changes within the context of a
We are aware of four commercial products, BIOSTER,
single input molecule, rather than listing literature
EMIL, WABE, and BIOISOSTER, developed indepen-
examples. The second commercial package, EMIL
dently, and possessing some features in common with
(example mediated innovation for lead evolution), com-
Drug Guru. Unfortunately, published scientific descrip-
bines a database analogous to the BIOSTER database
tions of each are limited or unavailable, precluding a de-
(3500 ‘optimization schemes' in the 2003 v.2.4 release)
tailed comparison. The following brief description is
with structure input and viewing that is analogous to
intended to draw attention to this area and to stimulate
Drug Guru.Unlike Drug Guru, the transformations
further research. The first commercial package, BIO-
are not collectively organized into rules-of-thumb writ-
STER (bioisosterism), is a database of pairs of mole-
ten in SMIRKS language, but rather each literature
cules differing in one structural element—termed
example of molecule pairs serves as a separate rule. Like
bioanalogous pairs in the original literature
Drug Guru, new transformations can be added to EMIL
A search of this database will generate literature exam-
via programming within the EMIL software. EMIL is
ples of pairs of compounds that illustrate many of the
installed as a stand-alone application (more recent ver-
same transformations encoded within Drug Guru. Rath-
sions are optionally web-based) in contrast to both BIO-
er than represent the changes as a list of general rules, as
STER (requires ISIS environment, Molecular Design
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
Figure 11. Examples of Drug Guru output for the input structure of sildenafil. The location of structural alteration is denoted with an arrow. Thetotal list contained 91 structures, and the arbitrary ranking within the list is designated here.
based scoring.The fourth commercial package, BIO-
ISOSTER, transforms an input structure according to
approximately 300 ‘target-specific' changes derived from
literature reports of kinase, protease, ion channel, phos-
phodiesterase, and nuclear receptor ligands.In addi-
tion to these commercial products, publications from
the Merck, Novartis, GlaxoSmithKline, Celera Genom-
ics, and Organon computational groups mention pro-prietary databases of functional group replacements
Figure 12. Chemical structure of vardenafil. This structure is created
and implementation within compound design pro-
by Drug Guru in a two-generation run using sildenafil as
input. The two generations use (1) the homologation-C rule, and (2)the NC-switch rule.
3.3. Limitations of this approach
Limited) and Drug Guru (requires Daylight toolkit and
From the listing of commercially available packages and
Oracle environments). A third commercial package,
publications discussed above, it is evident that molecular
WABE, generates isosteres of an input molecule accord-
replacement rule-based software for drug design is an
ing to an atom-replacement algorithm with an output
emerging and exciting area of research. In the case of
ranking based on electrostatic similarity or receptor-
Drug Guru, we feel that there are two main limitations
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
that happen to also be limitations common to other
computer-aided drug design programs: (1) imperfectranking scheme and (2) lack of synthetic chemistry
In this report, we describe a new web-based computer
knowledge. The first limitation is related to the age-old
application that applies a library of medicinal chemical
question of ‘how do you recognize a drug when you
transformation rules to an input structure and then per-
see it?' The example of the sildenafil analogs illustrated
mits evaluation of the resulting output structures. The
above is telling. As a chemist confronted with a list of
name of the new software captures this intellectual pro-
91 suggestions, how does one know, a priori, that num-
cess of drug generation using rules with the acronym
ber 64 (the vardenafil analog) is the structure on which
‘Drug Guru.' It is hoped that Drug Guru will provide
to focus attention? Also important is the question of
an intellectual guide to medicinal chemists in the
how good the remaining 90 structures are? Unequivocal
increasingly difficult task of discovering new compounds
answers to these questions are not possible. The main in-
as potential drug candidates.
tent of Drug Guru is to generate ideas. We submit thatthe idea-list that a medicinal chemist has in mind at anyone time is by its nature incomplete, and Drug Guru is
designed to help fill that gap. However, idea generationis not usually cited as the rate-determining step in drug
Funding for the Drug Guru project was provided by the
discovery. The ranking that Drug Guru currently pro-
vides is based on calculated physical properties and
Acknowledgment is also made to the Abbott Laborato-
has acknowledged weaknesses. We speculate that assess-
ries Medicinal Chemists who contributed many ideas for
ment of ‘success frequency' for individual rules may pro-
structural transformations (rules). Cheminformatics and
vide a novel way of ranking compounds for future
web programming was carried out by Moonview Con-
evaluation. Chemists could conceivably prioritize trans-
sulting, LLC.
formations that have historically performed well in cre-ating drug candidates, that is, transformations with thebest ‘yield.' Confounding any single prioritization
References and notes
scheme is the fact that analog design strategies at theoutset of a research project differ from those in a mature
1. Wermuth, C. G., Ed., The Practice of Medicinal Chemis-
program close to a clinical candidate. Currently in Drug
try; 2nd Ed.; Academic: New York, 2003.
Guru, all design rules are treated equally. We welcome
2. Sneader, W. Drug Prototypes and their Exploitation; John
suggestions on the optimal computational protocol for
Wiley & Sons: New York, 1996.
differentially weighting the rules.
3. Burger, A. Prog. Drug Res. 1991, 37, 287–371.
4. Chen, X.; Wang, W. Annu. Rep. Med. Chem. 2003, 38,
Assessing synthetic feasibility is an important part of
target evaluation. In an ideal setting within any comput-
5. Patani, G. A.; LaVoie, E. J. Chem. Rev. 1996, 96, 3147–
er-aided drug design software, the user would receive
quick feedback whether suggested output structures
6. Thornber, C. W. Chem. Soc. Rev. 1979, 8, 563–580.
7. Lipinski, C. A. Annu. Rep. Med. Chem. 1986, 21, 283–291.
were conveniently accessible from available starting
8. Spatola, A. Ann. Rep. Med. Chem. 1981, 16, 199–209.
materials, or would require multi-step syntheses with
9. Rudinger, J. In Drug Design; Ariens, J., Ed.; Academic:
varying degrees of difficulty and precedent. In fact, when
New York, 1971; Vol. 2, pp 319–419.
Drug Guru was evaluated with a test audience of 25
10. Drug Guru is a trademark of Abbott Laboratories. The
medicinal chemists, incorporation of synthetic chemistry
software is proprietary to Abbott Laboratories and
knowledge into the evaluation process was the single
Moonview Consulting, LLC. Readers desiring more
most requested improvement. Unfortunately, a fully sat-
information are encouraged to contact the first author.
isfactory computational protocol to conveniently, and
11. James, C. A.; Weininger, D.; Delaney, J., Daylight Theory
reliably, assess laboratory access to computer-generated
Manual, Daylight Chemical Information Systems, Inc.,
ideas is currently not possible. One simple, yet specula-
tive, possibility for Drug Guru would be to assign a ‘dif-
12. Jung, M. E.; Piizzi, G. Chem. Rev. 2005, 105, 1735–1766.
ficulty ranking' to each rule. Rules that correspond to a
13. Furet, P.; Bold, G.; Hofmann, F.; Manley, P.; Meyer, T.;
common laboratory operation, such as a methylation (the
Altmann, K.-H. Bioorg. Med. Chem. Lett. 2003, 13, 2967–
hydroxy-to-methoxy rule, rule 3, ), would be
ranked ‘high priority.' Rules that correspond to potential-
ly laborious multi-step procedures if interpreted literally,
15. Trejo, A.; Arzeno, H.; Browner, M.; Chanda, S.; Cheng,
such as ring transformations (for example, NC-switch,
S.; Comer, D. D.; Dalrymple, S. A.; Dunten, P.; Lafargue,
rule 10, would be ranked ‘low priority.' Subse-
J.; Lovejoy, B.; Freire-Moar, J.; Lim, J.; Mcintosh, J.;
quent sorting of the output list by the medicinal chemist
Miller, J.; Papp, E.; Reuter, D.; Roberts, R.; Sanpablo, F.;
would yield a list of the more synthetically accessible sug-
Saunders, J.; Song, K.; Villasenor, A.; Warren, S. D.;Welch, M.; Weller, P.; Whiteley, P. E.; Zeng, L.; Gold-
gestions high on the list. More extensive software links be-
stein, D. M. J. Med. Chem. 2003, 46, 4702–4713.
tween Drug Guru and organic chemistry synthesis design
16. Sildenafil patents US 5250534, US 5346901, US 5719283;
programs, such as Lhasa, Chiron, CAESA, etc, or reac-
vardenafil patents US 6362178, US 6566360.
tion databases, such as REACCS, can be envisioned. Re-
17. Representative references: docking/scoring programs: (a)
search into incorporating synthetic chemistry input into
Perola, E.; Walters, W. P.; Charifson, P. S. Proteins:
Drug Guru is in progress.
Struct. Funct. Bioinf. 2004, 56, 235–249; De novo pro-
K. D. Stewart et al. / Bioorg. Med. Chem. 14 (2006) 7011–7022
grams: (b) Stahl, M.; Todorov, N. P.; James, T.; Mauser,
21. Balakin, K. V.; Tkachenko, S. E.; Okun, I.; Skorenko, A.
H.; Boehm, H. -J.; Dean, P. M. J. Comput. Aided Mol.
V.; Ivanenkov, Y. A.; Savchuk, N. P.; Ivashchenko, A. A.;
Des. 2002, 16, 459–478; Scaffold hopping programs: (c)
Nikolsky, Y. Chem. Oggi 2004, 22, 15–18, The BIOISO-
Jenkins, J. L.; Glick, M.; Davies, J. W. J. Med. Chem.
STER program is commercially available from Chemical
2004, 47, 6144–6159; Pharmacophore programs: (d) Patel,
Diversity Labs, Inc.
Y.; Gillet, V. J.; Bravi, G.; Leach, A. R. J. Comput. Aided
22. Sheridan, R. P. J. Chem. Inf. Comp. Sci. 2002, 42, 103–108.
Mol. Des. 2002, 16, 653–681.
23. Ertl, P. J. Chem. Inf. Comp. Sci. 2003, 43, 374–380.
18. Ujvary, I. Pestic. Sci. 1997, 51, 92–95, The BIOSTER
24. Lewell, X. Q.; Jones, A. C.; Bruce, C. L.; Harper, G.;
program is commercially available from Accelry Inc.,
Jones, M. M.; Mclay, I. M.; Bradshaw, J. J. Med. Chem.
2003, 46, 3257–3274.
19. Fujita, T. In Trends in QSAR of Molecular Modeling 92;
25. Southall, N. T.; Ajay J. Med. Chem. 2006, 49, 2103–2109.
Wermuth, C. G., Ed.; ESCOM: Leiden, 1993; pp 143–159,
26. Wagener, M.; Lommerse, J. P. M. J. Chem. Inf. Model.
The EMIL program is commercially available from
2006, 46, 677–685.
CompuDrug International, Inc, .
27. Li, Q.; Chu, D. T. W.; Claiborne, A.; Cooper, C. S.; Lee, C.
20. Sayle, R., Skillman, G.; Nicholls, A., Abstracts, 227th
M.; Raye, K.; Berst, K. B.; Donner, P.; Wang, W.; Hasvold,
National Meeting of the American Chemical Society,
L.; Fung, A.; Ma, Z.; Tufano, M.; Flamm, R.; Shen, L. L.;
Anaheim, CA, March 28–April 1, 2004, American Chemical
Baranowski, J.; Nillius, A.; Alder, J.; Meulbroek, J.; Marsh,
Society: Washington, DC. The WABE program is com-
K.; Crowell, D.; Hui, Y.; Seif, L.; Melcher, L. M.; Henry, R.;
mercially available from OpenEye Scientific Software,
Spanton, S.; Faghih, R.; Klein, L. L.; Tanaka, S. K.;
Plattner, J. J. J. Med. Chem. 1996, 39, 3070–3088.
Source: http://iranarze.ir/wp-content/uploads/2016/06/3220-English-1.pdf
Checkpoint Contents Pension & Benefits Library Pension & Benefits Editorial Materials EBIA Benefits Compliance Library Cafeteria Plans XX. What Expenses Can Be Reimbursed Under a Health FSA? XX.M. Table of Common Expenses, Showing Whether They Are for "Medical Care" M. Table of Common Expenses, Showing Whether They Are for "Medical
JULY 2006 ISSUE Save The Date DRUGS USED TO TREAT BPH MAY September 14 ALSO PREVENT PROSTATE CANCER The KnowledgeNet "In TheKnow" Awards Luncheon by Diane Johnson New York, NYDetails at: ew evidence shows that doxazosin and terazosin (alpha-blockers), currently being used for the treatment of BPH (Benign Prostatic Hyperplasia) and hypertension, may alsodecrease the risk of developing prostate cancer. In addition, they may prevent the



