Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Exploiting the pleiotropic antioxidant effects of established drugs in cardiovascular disease
Int. J. Mol. Sci. 2015,
16, 18185-18223; doi:10.3390/ijms160818185
OPEN ACCESS
International Journal of
Molecular Sciences
ISSN 1422-0067
Exploiting the Pleiotropic Antioxidant Effects of Established
Drugs in Cardiovascular Disease
Sebastian Steven 1,2,†, Thomas Münzel 1,† and Andreas Daiber 1,†,*
1 2nd Medical Clinic, University Medical Center of the Johannes Gutenberg-University,
Mainz 55131, Germany; E-Mails:
[email protected] (S.S.);
[email protected] (T.M.)
2 Center for Thrombosis and Hemostasis, University Medical Center of
the Johannes Gutenberg-University, Mainz 55131, Germany
† These authors contributed equally to this work.
* Author to whom correspondence should be addressed; E-Mail:
[email protected];
Tel.: +49-(0)6131-17-6280; Fax: +49-(0)6131-17-6293.
Academic Editor: G. Jean Harry
Received: 26 June 2015 / Accepted: 27 July 2015 / Published: 5 August 2015
Abstract: Cardiovascular disease is a leading cause of death and reduced quality of life
worldwide. Arterial vessels are a primary target for endothelial dysfunction and
atherosclerosis, which is accompanied or even driven by increased oxidative stress. Recent
research in this field identified different sources of reactive oxygen and nitrogen species
contributing to the pathogenesis of endothelial dysfunction. According to lessons from the
past, improvement of endothelial function and prevention of cardiovascular disease by
systemic, unspecific, oral antioxidant therapy are obviously too simplistic an approach.
Source- and cell organelle-specific antioxidants as well as activators of intrinsic
antioxidant defense systems might be more promising. Since basic research demonstrated
the contribution of different inflammatory cells to vascular oxidative stress and clinical
trials identified chronic inflammatory disorders as risk factors for cardiovascular events,
atherosclerosis and cardiovascular disease are closely associated with inflammation.
Therefore, modulation of the inflammatory response is a new and promising approach in
the therapy of cardiovascular disease. Classical anti-inflammatory therapeutic compounds,
but also established drugs with pleiotropic immunomodulatory abilities, demonstrated
protective effects in various models of cardiovascular disease. However, results from
ongoing clinical trials are needed to further evaluate the value of immunomodulation for
the treatment of cardiovascular disease.
Int. J. Mol. Sci. 2015,
16
Keywords: cardiovascular disease; endothelial dysfunction; oxidative stress; inflammation;
dipeptidyl peptidase-4 inhibitors; glucagon-like peptide analogs
1. Oxidative Stress in Cardiovascular Disease and Inflammation
1.1. Introduction
The majority of cardiovascular diseases are accompanied by an imbalance between the formation of
reactive oxygen species (ROS, including superoxide, hydrogen peroxide as well as precursor products peroxynitrite or hypochlorous acid) and antioxidant enzymes [1,2], leading to a deviation from the steady state [3]. More recent evidence suggests that adverse redox signaling and oxidative stress are not only side effects of the progression of cardiovascular disease but even potent triggers of their development and pathogenesis [4,5]. According to the "kindling radical" hypothesis (or "bonfire" concept), the formation of ROS may trigger the activation of additional sources of ROS in certain disease conditions or during the aging process [6,7]. According to recent reports, vascular dysfunction in general, but hypertension and coronary artery disease may also be linked to inflammation or low-grade activation of the immune system [8,9]. Uncoupling of endothelial nitric oxide synthase (eNOS) is a hallmark of most cardiovascular disease [10,11] and endothelial dysfunction in coronary and peripheral vessels, measured by acetylcholine-dependent plethysmography or flow-mediated dilation, is an early predictor of cardiovascular events [12,13]. eNOS function is regulated by many different factors such as subcellular localization, calcium levels, binding of different co-factors (e.g., BH4, FAD, FMN, NADPH, zinc), and other proteins (e.g., calmodulin, heat shock proteins). Regulation of eNOS activity by so-called "redox switches" is of great interest for the present review—the oxidative depletion of tetrahydrobiopterin (BH4), oxidative disruption of the dimeric eNOS complex by oxidation of the zinc-sulfur complex,
S-glutathionylation of a cysteine in the reductase domain, and adverse phosphorylation at Thr495/Tyr657, as well as ROS-triggered increases in levels of the endogenous eNOS inhibitor asymmetric dimethylarginine (ADMA) (for detailed review see [6,7]). Another focus of research in the cardiovascular field is the "repair" of vascular damage by improvement of the function of endothelial progenitor cells by drugs with antioxidant and other pleiotropic properties [14] or infusion of these cells after a severe insult such as myocardial infarction [15,16]. This topic will only be touched on with some examples but not explored in detail. The major part of this review will discuss antioxidant therapeutic interventions that prevent eNOS uncoupling, thereby normalizing endothelial function in particular and improving cardiovascular disease in general. We will emphasize the importance of low-grade inflammation in the development of endothelial dysfunction and cardiovascular disease and discuss the contribution of specific inflammatory cells and their cytokine profiles to the development and progression of cardiovascular disease.
1.2. Inflammation, Oxidative Stress, and Endothelial Dysfunction
The first description of the role of oxidative stress in the development and progression of
cardiovascular disease in an experimental model of hypercholesterolemia was published by Harrison
Int. J. Mol. Sci. 2015,
16
and Ohara [17,18]. According to the abovementioned concept of "kindling radicals" or the "bonfire" hypothesis, the initial formation of superoxide (e.g., from phagocytic NADPH oxidases of infiltrated leukocytes) and subsequent formation of peroxynitrite most likely triggers further damage such as eNOS uncoupling, converting this beneficial nitric oxide synthase into a detrimental superoxide-producing enzyme (Figure 1) [6,8]. Likewise, ROS from infiltrated immune cells can activate or induce expression of vascular cell oxidases such as Nox1, Nox2, or Nox4 (isoform specific catalytic subunits of NADPH oxidases), or mediate the oxidative conversion of the xanthine dehydrogenase to the oxidase form [6,19]. This ROS-induced ROS formation is well known for cross-activation of mitochondrial ROS formation by dysfunctional mitochondria [20]. Mitochondrial ROS formation and release can be stimulated by thiol-oxidation in different mitochondrial structures (e.g., mitochondrial permeability transition pore constituents such as cyclophilin D, p66shc or monoamine oxidases); xanthine dehydrogenase is converted to the oxidase form by oxidation of critical thiol residues; the protective action of eNOS to produce •NO is switched to adverse superoxide formation by oxidative depletion of BH4, adverse phosphorylation by redox-activated kinases,
S-glutathionylation ,or oxidative disruption of the zinc-sulfur complex at the dimer binding interface, called the "uncoupling" process. These changes (increased vascular oxidative stress and release of inflammatory signaling molecules) will lead to endothelial cell activation and priming for the adhesion of additional immune cells as well as platelets and switch the vasodilatory, antiaggregatory, and antiatherosclerotic phenotype of the endothelium to a vasoconstrictory, proaggregatory, and proatherosclerotic one.
As described above, ROS formation is not only a side effect of cardiovascular diseases but directly
contributes to the disease progression in many ways. For example, the induction of endothelial dysfunction by oxidative modification of eNOS or its cofactors as well as redox stimulation of inflammatory cascades fosters the progression of these cardiovascular diseases (Figure 1). Most immune cells express high levels of functional NADPH oxidases and are capable of producing ROS at much higher levels than vascular cells [21,22]. The important role of phagocytic NADPH oxidase in this process was demonstrated by the fact that white blood cells with dysfunctional Nox2 were not able to infiltrate the vascular wall and induce vascular oxidative stress and damage [22,23], e.g., by oxidative conversion of xanthine dehydrogenase to the oxidase form, uncoupling of eNOS, or stimulation of mitochondrial ROS formation and release by specific redox switches [6]. On the other hand, there is clear evidence that ROS formation
per se contributes to a pro-inflammatory phenotype, since mitochondrial superoxide/hydrogen peroxide formation has the ability to activate immune cells [24–26]. ROS play an important role in inflammation and tissue damage [27]. There is also increasing evidence of a close interaction between vascular oxidative stress and inflammation during the aging process, leading to a vicious cycle in the aging vasculature [28]. By this crosstalk, infiltrated immune cells promote vascular oxidative stress, lead to endothelial cell activation, and prime the endothelium for the adhesion of additional leukocytes and platelets [8], which is of great importance for aging-associated endothelial dysfunction [29].
Vice versa, oxidative stress is a hallmark of all cardiovascular disease and will also lead to endothelial cell activation, priming for adhesion and infiltration of immune cells as well as activation of these infiltrated immune cells. Accordingly, most cardiovascular disease displays a low-grade inflammatory phenotype of the vasculature.
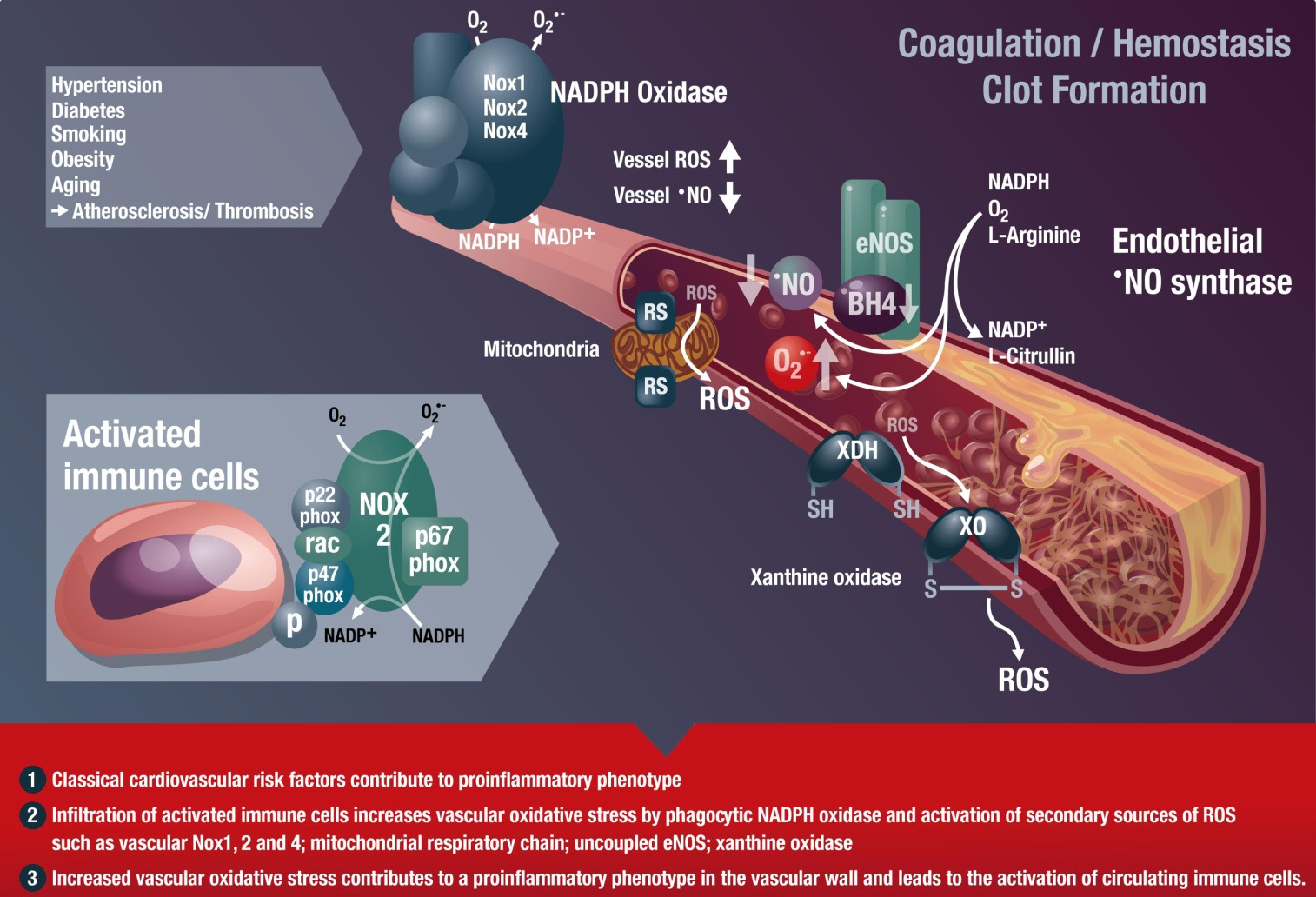
Int. J. Mol. Sci. 2015, 16
Figure 1. Inflammatory cells, vascular dysfunction, and atherothrombosis. The scheme
illustrates the activation of immune cells and recruitment to vascular tissues by classical
cardiovascular risk factors, leading to activation of secondary vascular ROS sources such
as NADPH oxidase (Nox1, Nox2, and Nox4), xanthine oxidase (conversion of the
dehydrogenase (XDH) to the oxidase (XO) form), mitochondria (via mitochondrial redox
switches (RS)), and uncoupled eNOS (oxidative depletion of tetrahydrobiopterin (BH4)
and other redox switches), all of which contribute to vascular dysfunction. Immune cells
such as monocytes need the functional phagocyte-type NADPH oxidase (Nox2) in order to
infiltrate vascular tissues. ROS produced by this activated Nox2 from infiltrated immune
cells will activate secondary vascular ROS sources in a redox-sensitive fashion (for review
see [19,30]). These processes lead to late-stage cardiovascular complications such as
atherosclerosis with plaque formation and thrombosis. Modified from [8]. With permission
by Bentham Science Publisher. Copyright 2014, Eureka Science Ltd.
Different immune cells have been reported to contribute to the development of cardiovascular
disease, but since they interact with each other, the individual impact of each cell type on the development of cardiovascular disease remains elusive. The contribution of B- and T-cells to the development of hypertension by angiotensin-II infusion was demonstrated by RAG-1−/− mice [22]. Likewise, the onset of angiotensin-II induced hypertension and vascular oxidative stress was also attenuated by removal of myelomonocytic cells [23]. Of note, immune suppressive treatment of patients with rheumatoid arthritis or psoriasis was associated with a reduction of systolic blood
Int. J. Mol. Sci. 2015, 16
pressure [31], underlining the clinical impact of a direct contribution of the immune system to vascular dysfunction.
1.3. Chronic Autoimmune Diseases Associated with Cardiovascular Disease
Chronic autoimmune diseases such as systemic lupus erythematodes, rheumatoid arthritis, and
severe psoriasis are associated with an increased risk for cardiovascular events [32–35]. Importantly, psoriasis was defined as an independent risk factor in addition to the classical cardiovascular risk factors such as smoking, obesity, and diabetes [36]. The European League against Rheumatism even recommends the management of cardiovascular risk in inflammatory arthritis in their guidelines [37]. As an early predictor of cardiovascular events, impaired vascular function was observed under chronic inflammation, which was evident in patients with rheumatoid arthritis by a significant increase in the intima-media thickness [38] or impaired endothelial function measured by flow-mediated dilatation in patients with psoriasis [39]. Therefore, recent clinical trials have demonstrated that increased cardiovascular mortality in patients with chronic inflammatory disease can be managed by targeting specific cytokines or activation of specific immune cells, e.g., in psoriasis the IL-17/IL-23 axis [40–42], in systemic lupus erythematodes IL-17A signaling [43], and in rheumatoid arthritis the IL-6, TNF-α, and IL-17A cascades [44,45]. These data provide a feasible link between cardiovascular disease and the chronic autoimmune diseases (also reviewed in [32,34]).
2. Classical Antioxidants and New Strategies to Modulate Oxidative Stress
2.1. Classical Antioxidants
Based on the oxidative stress concept in cardiovascular, neurodegenerative, metabolic, and
inflammatory disease [5,7,8], numerous studies were conducted. In vitro and animal studies of these diseases were performed in order to characterize the cytoprotective or therapeutic benefit of antioxidants and to promote phytochemicals, functional foods, and antioxidant (vitamin) supplements. However, antioxidants have failed to show any therapeutic benefit in most large clinical trials that were conducted according to modern standards [46], such as HOPE (Heart Outcome Prevention Evaluation) and HOPE-TOO (Heart Outcome Prevention Evaluation—The Ongoing Outcomes), which demonstrated that vitamin E causes more heart failure and left heart decompensation [47–50] (for review see [51]). A prospective study with vitamin C in post-menopausal women with diabetes mellitus even demonstrated an increased incidence of cardiovascular events and
mortality under antioxidant therapy [52]. The SAINT I trial investigated the therapeutic benefit of the synthetic antioxidant, NXY-059, in acute ischemic stroke but failed to show any neuroprotective effect [53]. According to Bjelakovic and coworkers, meta-analysis of 68 randomized trials with 232,606 participants revealed that the use of lipid-soluble antioxidants without medical indication may even increase mortality in adults [54]. Another meta-analysis of 14 randomized trials with 170,525 individuals by the same author demonstrated a similar trend—lipid-soluble antioxidants increased the mortality of gastrointestinal cancer patients [55]. However, other meta-analyses support the beneficial effects of vitamin C in specific disease conditions or disease-associated impairment of
Int. J. Mol. Sci. 2015, 16
functional parameters, e.g., on the survival of women with breast cancer [56] or on endothelial function in patients with atherosclerosis, diabetes, and heart failure [57].
These large-scale clinical trials on chronic oral antioxidant supplementation are contrasted by
multiple small cohort studies with acute (parenteral) administration of antioxidants with highly beneficial effects on the surrogate parameters of disease (e.g., endothelial dysfunction) in chronic smokers or patients with diabetes or coronary artery disease [12,58–60] (for review see [51]). The advantage of parenteral administration of water-soluble antioxidants is that high plasma concentrations of the antioxidant are achieved [61], thereby omitting the complications of oral absorption (time-lag, limited capacity) and insufficient compliance. High-dose intravenous infusion of vitamin C also improved endothelial function in patients with Kawasaki disease [62], kidney dysfunction [63], hypertension [64], liver cirrhosis, and portal hypertension [65]. Moreover, parenteral application of vitamin C has also proven to have clinical effects in patients with allergies [66], sudden hearing loss [67], breast cancer, infection, and pancreatitis [68].
A positive example of the beneficial effect of chronic antioxidant therapy is vitamin D. Lack of
vitamin D is endemic in the human population and epidemiological data indicate that deficiency of this vitamin is associated with cardiovascular disease [69]. There is some evidence from interventional trials demonstrating that supplementation of vitamin D is beneficial to endothelial function [70,71], blood pressure [72], and cardiac hypertrophy [73,74] in humans. Furthermore, a recent Cochrane analysis revealed that vitamin D supplementation significantly reduces cardiovascular mortality in elderly people [75]. Nevertheless, further large-scale randomized placebo-controlled clinical trial are needed to elucidate the cardiovascular protective effects of vitamin D. In contrast to other vitamins, deficiency of vitamin D is very common, especially in older individuals [76], which might be the explanation for the beneficial effects of vitamin D, especially on cardiovascular disease in the elderly [77].
Based on the disappointing results of most large-scale clinical trials (HOPE, HOPE-TOO) [47,49]
(reviewed in [51]), with chronic oral antioxidants supplementation the question arises whether the oxidative stress hypothesis in pathogenesis and disease progression is wrong? We think the answer is no and possible explanations for the lack of clinical efficacy of antioxidants in these studies might be that: (1) vitamins C and E act as pro-oxidants; (2) the coronary artery disease of the patients included in the studies is already irreversible; (3) the coronary artery disease patients are already being treated with drugs displaying antioxidant properties (for example, ACE inhibitors and angiotensin type 1 receptor blockers; see also Section 3); (4) chronic antioxidant therapy inhibits intrinsic ischemic preconditioning, which relies on ROS formation; and (5) oral vitamin treatment does not result In high enough concentrations of the antioxidants at the place of oxidative stress (summarized from [5]). Another important drawback of classical antioxidants may be the slow reaction between O •−
vitamins C and E (with rate constants of 3.3 × 105 and 4.9 × 103 M−1·s−1, respectively, compared with 1.9 × 1010 M−1·s−1 for the reaction between •NO and O •−
2 ) [78]. Finally, in most of the abovementioned
large-scale clinical trials on the use of chronic oral antioxidant supplementation, the compliance of the patients was not controlled (e.g., by measurement of plasma levels of the antioxidants). The important role of controlled antioxidant plasma levels became evident in the EPIC Norfalk study, demonstrating that vitamin C concentrations in the blood inversely correlate with all-cause mortality in healthy volunteers [79]. In addition, there was an inverse correlation between circulating vitamin C concentrations
Int. J. Mol. Sci. 2015, 16
and risk of stroke as reported by a meta-analysis [80]. According to a concept put forward by Lykkesfeldt and colleagues [81], better results of antioxidant therapy might be expected under conditions of antioxidant deficiency (e.g., for vitamin C and E) [82], or often encountered for vitamin D [76]. Some of these reasons would favor a proper prediagnosis of patients for blood levels of antioxidants, strict monitoring of this parameter during antioxidant therapy, and acute infusion of vitamin C in accordance with the observations and advantages of parenteral use discussed above. According to a recent review on the use of antioxidants in translational medicine, future antioxidant strategies will not be based on the classical antioxidant vitamins (apart from some acute situations with subclinical deficiencies as well as parenteral instead of oral therapy) but rather on the activation of endogenous antioxidant enzyme systems, inhibition of critical ROS sources (e.g., NADPH oxidase), the repair of oxidatively damaged structures, or site-directed antioxidant approaches [51].
2.2. New Antioxidant Strategies
Direct and cell organelle-specific targeting of ROS formation is a new promising strategy. A
prominent example is scavenging mitochondrial ROS by mitochondria-targeted antioxidants such as mitoquinone (mitoQ) [83,84], a quinone that is coupled to a triphenylphosphonium group to facilitate mitoQ accumulation in mitochondria by up to 10,000-fold. MitoQ showed beneficial effects in an animal model and in human cells from patients with chronic obstructive pulmonary disease [85], improved nitrate-tolerance-associated side effects of nitroglycerin therapy in rats [86], and normalized endothelial function and cardiac hypertrophy in stroke-prone spontaneously hypertensive rats [87]. Moreover, MitoQ showed neuroprotective effects in experimental amyotrophic lateral sclerosis [88], suppressed NLRP3 inflammasome-mediated inflammatory cytokines in a murine colitis model [89], beneficially influenced nephropathy in diabetic mice [90], and prevented cardiac ischemia-reperfusion injury in rats [84]. According to recent data, another mitochondria-targeted antioxidant, mitoTEMPO, prevented adverse effects of angiotensin-II in experimental hypertension [91]. Similar beneficial effects have been reported for the use of mitochondria-targeted SOD mimetics such as
Mn(III) 5,10,15,20-tetrakis(N-methylpyridinium-2-yl)porphyrin (MnTM-2-PyP5+) in various disease models [92,93]. Several compounds of the class of mitochondria-targeted antioxidants are currently in late phase clinical trials (for review see [92–95]; for ongoing clinical trials visit www.clinicaltrials.gov). A limitation of the use of mitochondria-targeted antioxidants might be that viable mitochondria with intact membrane potential are required for their mitochondrial accumulation, which could interfere with their uptake, especially in dysfunctional, ROS-producing mitochondria. A similar idea provides the basis for endothelium-targeted antioxidants. Shuvaev et al. demonstrated that targeting of SOD, but not catalase, to the endothelium reversed angiotensin II-induced endothelial dysfunction [96], nicely demonstrating that O •−
2 is a more harmful species in the vascular system
than H2O2. The antioxidant enzymes SOD and catalase were conjugated with an antibody against PECAM-1 (platelet/endothelial cell adhesion molecule-1) to ensure endothelial binding. Another strategy would be to covalently bind SOD mimetics or antioxidants to heparin [97], which will lead to the binding of these antioxidant compounds to heparin-binding sites on the endothelial cell layer [98,99].
Modulation/regulation of endogenous antioxidant defense systems or ROS sources by miRNAs
(e.g., antagomirs), epigenetic drugs (e.g., modulators of histone acetyl transferases or deacetylases),
Int. J. Mol. Sci. 2015, 16
or phytochemicals are other completely new antioxidant strategies [100–106]. Resveratrol, a phytochemical antioxidant, was previously regarded as a direct ROS scavenger, but more recent data revealed that it mostly acts via indirect antioxidant mechanisms [107], e.g., by modulation of gene expression via miRNAs, epigenetic modifications, and direct effects on proteins of the DNA repair machinery [108–110]. Besides resveratrol, there are hundreds of these phytochemical antioxidant compounds, in many cases with proven therapeutic effects and in some cases even with mechanistic explanations for these observed clinical effects. One prominent example is Ginkgo biloba, which has been in clinical use for a long time, especially for dementia therapy but also for its positive effects on cardiovascular disease [111].
Other antioxidant strategies are only briefly mentioned here (we refer to the respective review articles,
e.g., [51]) and are based on: (1) the inhibition of disease-relevant ROS sources such as inhibitors of NADPH oxidase (Nox) enzymes [112–115] or xanthine oxidase [116,117]; (2) upregulation of the endogenous antioxidant defense system such as Nrf2 agonists [118]; and (3) repair of oxidatively damaged protein structures as exemplified by activators of heme-deficient or oxidized soluble guanylyl cyclase [119–121]. According to Stocker and colleagues, an antioxidant should ideally be recycled by cellular reducing systems, act catalytically to prevent its consumption, or induce endogenous antioxidant defense systems rather than act as a direct scavenger (for review see [122]).
3. Antioxidants 2.0—Pleiotropic Antioxidant Effects of Established Drugs
It would be beyond the scope of this review to discuss all of the important representatives of this
group of drugs, so the following will be limited to some examples of new function for old drugs. The pleiotropic antioxidant effects of these compounds are characterized by different modes of action (as already described above): (1) induction of intrinsic antioxidant systems; (2) inhibition of Nox2-dependent ROS formation; and (3) direct ROS scavenging activity. It remains to be established whether some of these pleiotropic antioxidant effects are just a consequence of the primary pharmacological action of the drugs (e.g., lowering of blood pressure). At least in some of the examples the primary pharmacological action of the drugs can be excluded since data were obtained either in cell culture or even with isolated enzymatic systems.
3.1. Statins, ACE-Inhibitors, and AT1-Receptor Blockers
Angiotensin-converting enzyme inhibitors, type 1 angiotensin II receptor antagonists, statins, and
many other cardiovascular drugs display pleiotropic indirect antioxidant properties (e.g., inhibition of Nox enzymes and secondary to this prevention of eNOS uncoupling) [123,124]. In more detail, angiotensin-converting enzyme inhibitors and type 1 angiotensin II receptor antagonists increase the bioavailability of •NO by decreased breakdown of bradykinin and activation of the corresponding B2 receptor [125]. They also prevent the activation of the phagocytic and vascular Nox2 enzyme and thereby decrease cellular superoxide, hydrogen peroxide, and peroxynitrite levels [126]. Inhibition of angiotensin-II signaling decreases oxidative stress since angiotensin-II via its receptor leads to the formation of diacylglycerol, a potent endogenous trigger of NADPH oxidase activity [127]. In addition, these drugs confer potent anti-inflammatory effects by interfering with the adhesion of monocytes to the endothelium [128] and even improving the severity of adjuvant arthritis [129]. In
Int. J. Mol. Sci. 2015, 16
summary, angiotensin-converting enzyme inhibitors and type 1 angiotensin II receptor antagonists promote a vasodilatory, antithrombotic, and antiproliferative milieu and improve the function of endothelial progenitor cells [130]. These protective mechanisms might also explain their benefit for the therapy of patients with heart failure [131]. Statins obviously target exactly the same pathophysiological parameters. In patients with cardiovascular disease, statins reduce vascular inflammation and atherothrombosis, which causes cardiovascular events like myocardial
infarction [132–134]. Statins reduce NADPH oxidase activity in a Rac1-dependent mechanism [135,136] and improve the bioavailability of •NO through increased levels of the eNOS cofactor BH4, decreased levels of the endogenous eNOS inhibitor asymmetric dimethyl-L-arginine, decreased caveolin-1 activity, improved activating eNOS phosphorylation, and upregulation of eNOS mRNA [137]. The beneficial pleiotropic effects of statins are probably based on the induction of the Nrf2-heme oxygenase-1 system [138,139] but improvement of the function of endothelial progenitor cells could also contribute to their protective profile [140].
3.2. Nebivolol, Hydralazine, and Pentaerythrityl Tetranitrate (PETN)
One of the first known antihypertensive drugs was hydralazine, which is today mainly used for the
treatment of pre-eclampsia [141]. However, it experienced a "revival" when the company NitroMed introduced their combination drug BiDil containing hydralazine and isosorbide dinitrate. This combination therapy showed an impressive decrease in mortality in African-Americans with severe heart failure, who responded poorly to ACE inhibitors and other standard medications (the study design was based on data from V-HeFT (Vasodilator Heart Failure Trial) and A-HeFT (African-American Heart Failure Trial) [142–144]. According to our previous observations, hydralazine is a highly efficient peroxynitrite scavenger and prevents tyrosine nitration [145,146], which may at least contribute to its beneficial effects on nitroglycerin-induced nitrate tolerance [147] and potentially isosorbide dinitrate-associated side effects. Based on these data, we postulate a direct antioxidant property of hydralazine by scavenging peroxynitrite, a potentially harmful oxidant. This provides the rationale for the beneficial effects of the hydralazine/isosorbide dinitrate combination to prevent side effects of the organic nitrate under chronic therapy (e.g., endothelial dysfunction [148]). Recent data support a reaction between peroxynitrite and dihydralazine sulfate [149]. However, there is also evidence for the indirect antioxidant effects of hydralazine by induction of hypoxia-inducible factor-1α, vascular endothelial growth factor, and angiogenesis by inhibition of prolyl hydroxylases [150]. Among other antihypertensive drugs, hydralazine has been found to possess pleiotropic antioxidant effects in patients beyond the direct blood pressure lowering effects [151]. Also, protective effects of hydralazine on endothelial progenitor cell function were reported [152].
The third generation beta-blocker nebivolol was reported to induce vascular nitric oxide formation
via stimulation of eNOS activity in ex vivo studies [153–155], providing the rationale for improved •NO bioavailability in patients with essential hypertension [156]. In this clinical study a combination therapy of nebivolol/bendrofluazide, in contrast to atenolol/bendrofluazide treatment, improved •NO bioavailability despite a similar degree of blood pressure lowering. We could detect eNOS-stimulating effects of nebivolol neither in cultured endothelial cells nor in hypertensive mice when comparing wild-type controls with eNOS knockout mice (unpublished data, Karbach et al. and Daiber). We have
Int. J. Mol. Sci. 2015, 16
previously shown that nebivolol, in contrast to metoprolol and atenolol, prevents eNOS uncoupling and induction of phagocytic NADPH oxidase activity in white blood cells, vascular oxidative stress, and endothelial dysfunction in hyperlipidemic Watanabe (WHHL) rabbits [157]. In a subsequent study we characterized nebivolol as a potent Nox2 inhibitor in hypertensive rats as well as isolated cells, which are not shared by first- and second-generation beta-blockers [158]. Nebivolol directly interferes with the assembly of Nox2 and cytosolic subunits p47phox, p67phox, and rac1 in the cytoplasmic membrane, suggesting that Nox2 inhibition leads to reduced superoxide formation, prevents eNOS uncoupling and breakdown of nitric oxide by reaction with superoxide, and finally ameliorates endothelial function. This concept goes hand in hand with human data in which nebivolol normalized oxidative stress in hypertensive patients and led to reduced oxidative degradation of nitric oxide [159]. Finally, nebivolol improved the function of early endothelial progenitor cells in experimental myocardial infarction, which could also contribute to its beneficial clinical profile [160].
After the development of pentaerithrityl tetranitrate (PETN) for the U.S. market, it was abandoned,
but then used for many years in the former Eastern German Republic. After the reunion of Germany, PETN became the best-selling nitrate on the German market. PETN is the only organic nitrate in clinical use devoid of induction of nitrate tolerance, endothelial dysfunction, and other nitrate-associated side effects in volunteers [161,162] and patients with coronary artery disease [163,164]. The molecular explanation for the beneficial effects of PETN, not shared by other nitrates, is the induction of heme oxygenase-1 [165–168] in a Nrf2-dependent fashion [169]. PETN also induced extracellular superoxide dismutase [170], prevented vascular complications in experimental diabetes and hypertension [165,169], prevented the progression of atherosclerosis in a rabbit model [171], and inhibited platelet aggregation in heart failure [172]. In contrast to other organic nitrates, PETN improved the function (migration and incorporation) of endothelial progenitor cells and decreased their NADPH oxidase activity ex vivo and in vivo in humans and rats [173–175]. In addition, PETN therapy leads to the regulation of more than 1200 genes and upregulates several cardio-protective transcription factors, whereas nitroglycerin, also a nitrovasodilator, regulated approximately 500 genes different from those regulated by PETN [176]. More recently, PETN was shown to induce heritable epigenetic changes envisaged by H3K27 acetylation, H3K4 trimethylation, and transcriptional activation of eNOS, MnSOD, glutathione peroxidase-1, and heme oxygenase-1, all of which lead to reduced blood pressure in female offspring of PETN-treated hypertensive rats [177]. Of note, these beneficial effects were neither shared by other organic nitrates nor by the •NO donors tested in this study. The ongoing CAESAR trial (Clinical efficacy study of Pentalong for pulmonary hypertension in heart failure; EudraCT Number: 2009-015059-26) will show whether the potent antioxidant and vasculoprotective effects of PETN can be translated to patients with pulmonary hypertension as a result of heart failure.
3.3. Gliptins and Glucagon-Like Peptide-1 (GLP-1) Analogs Display Antioxidant and Anti-Inflammatory Properties
Dipeptidyl peptidase-4 (DPP-4) is an exopeptidase also known as CD26. N-terminal dipeptides are
cleaved from alanine- and proline-rich proteins [178]. Besides DPP-4, the family of DPPs consists of several members: DPP-1–DPP-4, DPP-6–DPP-9, quiescent cell proline dipeptidase (QPP), and fibroblast activation protein (FAP) [178,179]. DPP-4 has a wide range of functions, the best
Int. J. Mol. Sci. 2015, 16
characterized of which is the degradation of incretins (glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide GIP) [179]. Furthermore, DPP-4 cleaves non-incretin peptides, possesses non-enzymatic function, and interacts with membrane bound proteins as a chaperone [179]. In various tissues DPP-4 is expressed on the surface of endothelial cells, epithelial cells, and inflammatory cells (monocytes, lymphocytes, dendritic cells, and natural killer (NK) cells) [180–183].
GLP-1 is an incretin hormone released from L-cells in the intestine after food uptake [184,185].
In the context of glucose homeostasis, circulating GLP-1 binds to its receptor, which is expressed on pancreatic beta-cells, but also on cardiomyocytes, endothelial cells, and inflammatory cells. The GLP-1 receptor belongs to the family of G-protein-coupled receptors. After binding of GLP-1 to its receptor, cAMP levels rise and insulin release is stimulated. On pancreatic alpha-cells, GLP-1 reduces glucagon release (for review see [186]). In summary, GLP-1 is involved in glycemic control, which makes it an attractive target for treatment of diabetes [187,188]. Derived from the prolucagon gene, GLP-1 (7–36-amide) and GLP-1 (7–37) are secreted. Due to rapid degradation of GLP-1 to GLP-1 (9–36-amide) by DPP-4, the half-life of GLP-1 is below 2 min [189,190]. There are two pharmacological strategies for using the GLP-1 effects on glucose metabolism in diabetic patients: (1) inhibition of DPP-4 by gliptins to increase GLP-1 levels and (2) supplementation of modified GLP-1, which resists degradation by DPP-4. At the time of this review five DPP-4 inhibitors are approved by the European Medicines Agency (EMA) (vildagliptin, alogliptin, sitagliptin, linagliptin, and saxagliptin) for treatment of type 2 diabetes mellitus, also reflected by the Global Guideline for Type 2 Diabetes [191]. GLP-1 analogs are represented by liraglutide and exenatide, also reflected by the Global Guideline for Type 2 Diabetes [191].
Besides their potent effects on glycemic control in diabetic patients, research of the last years
revealed their effects on several other cell types and tissues. In vivo and in vitro studies demonstrated the beneficial effects of DPP-4 inhibitors on cardiovascular disease [192,193], but also in diseases like psoriasis [194], hepatic steatosis [195], or stroke [196]. Interestingly, pathogenesis of all of these diseases has oxidative stress and most likely inflammation in common. Finally, DPP-4 inhibitors improved the number and "homing" of endothelial progenitor cells in animal [197,198] and human [199] studies, but the number of publications on this association is still low. GLP-1 analogs seem to share these effects on endothelial progenitor cells [200].
Oeseburg et al. published evidence for a protective effect of DPP-4 inhibition on oxidative
stress-induced DNA damage and cellular senescence in Zucker diabetic fatty rats [201]. The authors propose elevated GLP-1 to be responsible since the effect could be blocked by exendin fragment 9–39, which is a GLP-1 receptor antagonist. According to this study, induction of the antioxidant enzymes heme oxygenase-1 and NADPH dehydrogenase (quinone) via protein kinase A activation are responsible for the beneficial effects [201]. The mentioned study is an excellent example of the difficulty of differentiating between the direct GLP-1 effects and the GLP-1-independent effects of DPP-4 inhibition. Others detected reduced oxidative stress under DPP-4 inhibitor therapy in animal models of type 1 diabetes [202], cardiac ischemia/reperfusion-injury [203], chronic myocardial infarction [204], abdominal aortic aneurysm [205], Parkinson's diseases [206], and sepsis [207,208]. Furthermore, limited data are available on the reduction of oxidative stress by DPP-4 inhibition in humans. Shah et al. found reduced 3-nitrotyrosine levels in isolated human pancreatic cells after treatment with linagliptin [209], which agrees with the findings in gliptin-treated type 2 diabetic
Int. J. Mol. Sci. 2015, 16
patients [210]. In humans it remains unclear whether DPP-4 inhibitor-dependent reduction of oxidative stress is independent of glucose-lowering effects. However, there is clear evidence for glucose-independent reduction of oxidative stress by DPP-4 inhibition in different animal models.
DPP4 inhibition has been shown to reduce oxidative stress in various disease models. Reports on
diabetes [211,212], atherosclerosis [192,193], sepsis [207,208], and neurological disease [213] can be found in the literature. AMP-activated protein kinase (AMPK) is an important regulator of oxidative stress in the vasculature, more specifically in endothelial cells [214]. Activation of AMPK via GLP-1 receptor signaling has been shown to reduce oxidative stress in cardiomyocytes and reduces activation of NADPH oxidase [215]. On the other hand, suppression of protein kinase C (PKC)/NFκB-dependent Nox activation/upregulation might also be responsible [211,212]. For GLP-1 independent action of DPP-4 inhibition on reduction of oxidative stress, it has been proposed that DPP-4 is an adenosine deaminase (ADA)-binding protein and regulates the subcellular localization and activity of this enzyme, which has known immunomodulatory functions [216,217]. ADA activity also leads to increased inosine levels with subsequent hypoxanthine formation and thereby provides the substrate for the pro-oxidative enzyme xanthine oxidase (XO) [208]. Furthermore, several other protein targets were described for DPP-4 such as caveolin-1, kidney Na+/H+ ion exchanger 3, thromboxane A2 receptor, CXCR4, CXCL12 (SDF-1), fibronectin, and many more [179], most of them being involved in the regulation of inflammation. Immunomodulation by DPP-4 seems to be critical for antioxidant properties of DPP-4 inhibitors.
Since various cell types and tissues are affected by DPP-4 inhibition and also by GLP-1, it is
difficult to determine which signaling pathway is predominantly responsible for reduction of oxidative stress in a specific disease model. Most of the studies that investigated the effects of DPP-4 inhibition on oxidative burst performed no experiments with genetic or pharmacological inhibition of the GLP-1 receptor. This limitation prevents a differentiation between DPP-4- and GLP-1-dependent effects. Future studies on cell-specific GLP-1 receptor knock-out animals are needed to draw a clearer picture of the complex interaction of DPP-4 and GLP-1. The following sections will focus on the antioxidant effects of DPP-4 inhibition and GLP-1 analog supplementation in atherosclerosis and sepsis.
3.3.1. Gliptins and GLP-1 in Atherosclerosis
As described above, cardiovascular diseases are closely linked to oxidative stress and inflammation.
Endothelial function is a reliable predictor of future cardiovascular events and is directly linked to the burden of oxidative stress in the vessel wall as well as in the blood (e.g., activation state of circulating immune cells) [12]. Recent studies demonstrate that walking distance and critical limb ischemia correlate with the activation state and production of ROS of circulating leukocytes in patients with peripheral artery disease [218–220]. Impaired endothelial function leads to inadequate vasodilation and increased disposition for infiltration of inflammatory cells. There is convincing evidence that atherosclerosis can be regarded an inflammatory disease [221].
Matsubara et al. investigated the effects of the DPP-4 inhibitor sitagliptin in an animal model of
atherosclerosis [192]. They used ApoE−/− mice on a high-fat diet. Sitagliptin treatment reduced atherosclerotic lesions, improved endothelial function, and reduced infiltration of CD68+ cells into the
Int. J. Mol. Sci. 2015, 16
vascular wall. Vascular inflammation was significantly reduced by sitagliptin treatment, which was proven by reduced mRNA levels of several pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α). Similar reduction of inflammation was found in cultured human monocytes. The authors also showed the anti-inflammatory effects of a GLP-1 analog (envisaged by reduced IL-6), which was additive to the beneficial effects of sitagliptin. This in vitro experiment demonstrated that the anti-inflammatory effects of DPP-4 inhibitor and GLP-1 analog were not connected to each other. Shah et al. made similar observations in LDLr−/− and ApoE−/− mice by using alogliptin treatment [193]. They demonstrated nicely the reduction of chemotaxis and monocyte activation by DPP-4 inhibitor therapy in both models of atherosclerosis. A major limitation of the study is that it does not differentiate between GLP-1- and DPP-4-dependent effects [193]. Besides the beneficial effects of DPP-4 inhibition on atherosclerosis, anti-atherosclerotic effects of GLP-1 supplementation were also demonstrated in animal models. GLP-1 therapy reduced vascular inflammation and increased plaque stability [222]. Others demonstrated improved endothelial function in ApoE−/− mice [223]. Since oxidative stress, derived from inflammatory monocytes, is a major trigger for endothelial dysfunction [23], the beneficial effects of GLP-1 in this context might rely on inhibition of this cell type. This hypothesis is supported by a recent publication reporting on reduced oxidative stress in human monocytes after exendin-4 incubation [224]. Furthermore, the antioxidant capacity (superoxide dismutase activity) in these cells was increased by exendin-4, which could be blocked by the PKA inhibitor H89 [224]. Others attributed the anti-inflammatory effects of GLP-1 to the modulation of NFκB-activity via PKA-dependent signaling pathways [225]. GLP-1 supplementation and DPP-4 inhibition induce protective effects on vascular function in animal models of atherosclerosis. Both reduce vascular inflammation, a main trigger for oxidative stress in the vasculature. Since the GLP-1 receptor and DPP-4 are expressed in endothelial cells, it may be suggested that both can improve endothelial function by direct effects. Indeed, improved function of the endothelial NO synthase in response to GLP-1 analog treatment was demonstrated, followed by reduced activation of endothelial cells via the PI3 kinase/Akt-signaling pathway [226]. Likewise, an activation of the cAMP/PKA-signaling pathway but also the cGMP-signaling pathway was reported for GLP-1 analogs [227,228]. Also, for the DPP-4 inhibitor alogliptin, potent vasodilatory effects were described based on a Src-Akt-eNOS-dependent nitric oxide release [229]. Similar vasodilatory effects were described for linagliptin in an eNOS and soluble guanylyl cyclase-dependent fashion [208]. Furthermore, studies with HUVECs revealed the inhibitory effects of GLP-1 analog treatment on mRNA expression of Nox subunits gp91 an p22phox [230].
Future studies and the use of cell-specific knock-out animals (DPP-4 and GLP-1 receptor) are
needed to differentiate between the DPP-4- and GLP-1-dependent effects on vascular oxidative stress and inflammation in models of atherosclerosis.
3.3.2. Gliptins and GLP-1 in Sepsis and Chronic Inflammatory Disease
Sepsis is an inflammatory disease that affects the whole organism. Depending on the immune status
of a patient, simple pneumonia can expand to a "systemic inflammatory response syndrome" (SIRS), which is a severe, life-threatening condition. An average mortality of 40% makes it a leading cause of death in the European Union [231]. Because of antibiotic resistance, the use of invasive procedures,
Int. J. Mol. Sci. 2015, 16
and an aging population, sepsis has become more frequent and the absence of efficient causal therapies confers high importance and priority to future research on the septic pathomechanisms but also on more promising therapeutic interventions [232].
Endotoxins (e.g., lipopolysaccharide, LPS) are responsible for the pathogenesis of sepsis in
humans and animals. They are part of the outer membrane of gram-negative bacteria and trigger the pathophysiological effects (e.g., circulatory disorders). Hypotension, impaired oxygen utilization, lactic acidosis, and aggravated blood flow in the microcirculation are characteristic of sepsis, by which multiple organ failure is caused [233–235]. Previous studies (animal and human) could show a correlation between sepsis and endothelial dysfunction, in which oxidative stress and endothelium-derived mediators (e.g., NO, prostacyclin) are involved [236–238]. The oxidative burst is a key feature of neutrophils and monocytes/macrophages, which play a pivotal role in host defense. NADPH oxidase isoform 2 is generating superoxide anion radicals in response to stimuli like LPS (gram-negative bacteria) or zymosan-A (fungi). Chronic granulomatous disease (CGD) underlines the importance of Nox2-derived superoxide anion radicals for host defense. In these patients Nox2 is dysfunctional because of a genetic mutation of the gp91phox gene and they are highly susceptible to infections [239]. Studies on critically ill patients revealed the protective effects of antioxidant therapy with ascorbate and α-tocopherol [240]. It reduced the risk of organ dysfunction and duration of hospitalization in these patients [240], whereas studies on other antioxidants revealed no beneficial effects [241]. The reason for these conflicting results might be that ROS formation is needed for host defense against bacteria.
A global reduction of oxidative stress in sepsis seems not to be the perfect solution to improve
survival. The most promising strategy would reduce the overshooting inflammatory response in septic patients but leave the basal host defense intact. Therefore, the question is whether systemic anti-inflammatory therapy of sepsis could counteract the overshooting immune response and might improve survival. The CORTICUS trial investigated whether hydrocortisone application, as a general suppressor of inflammation, might improve survival in septic patients. In this trial no significant improvement of survival in patients suffering from septic shock was found [242]. Furthermore, statins have anti-inflammatory properties and a recent trial tested the use of rosuvastatin in patients suffering from acute respiratory distress syndrome (ARDS). The results were disappointing and statin therapy failed to improve clinical outcome [243].
The immunomodulatory actions of DPP-4 inhibitors and GLP-1 analogs have already been
discussed in general and for models of atherosclerosis. Our group investigated the anti-inflammatory and antioxidant effects of DPP-4 inhibition and GLP-1 supplementation in models of endotoxemia. For induction of endotoxemia, the LPS injection model (10 mg/kg) was used, whereas a higher dose of LPS (17 mg/kg) was used to induce partial mortality. In a first study, we were able to show that endothelial dysfunction was severely impaired in endotoxemic rats [208]. This finding was associated with increased oxidative stress in whole blood, vessel walls, and hearts of the animals. Oral treatment with the DPP-4 inhibitor linagliptin strongly improved endothelial function and accordingly reduced oxidative stress levels. Interestingly, the reduced oxidative stress in the vascular wall was accompanied by less infiltration of CD11b+ cells and reduced myeloperoxidase protein expression. Our results revealed two different mechanisms for reduced oxidative stress under endotoxemic conditions in the vascular wall: (1) linagliptin treatment prevents expression of leukocyte adhesion molecules like
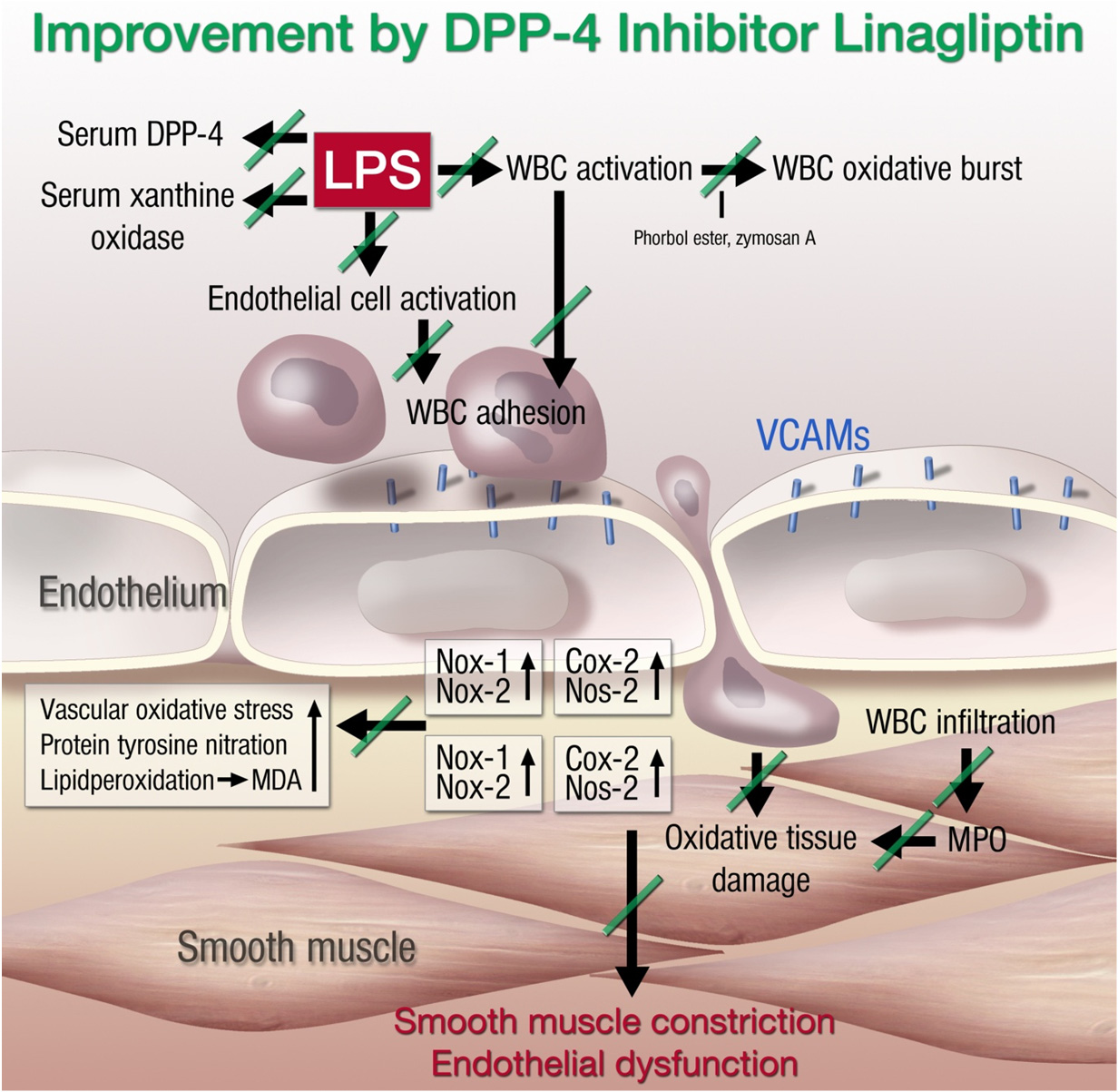
Int. J. Mol. Sci. 2015, 16
vascular adhesion molecule-1 (VCAM-1) and thereby reduces endothelial cell activation; and (2) linagliptin directly reduces LPS-induced activation of polymorphonuclear neutrophils (PMN), which was reflected by attenuated adhesion to endothelial cells and oxidative burst. In summary, DPP-4 inhibition reduces the inflammatory state of circulating leukocytes as well as the pro-inflammatory phenotype of the vascular wall and improves the function of endothelial cells, all of which ameliorates vascular function under endotoxemic conditions (Figure 2).
Figure 2. Proposed mechanisms of lipopolysaccharide (LPS)-induced vascular dysfunction
and improvement by linagliptin therapy. LPS treatment activates white blood cells (WBC,
envisaged by increased oxidative burst), increases serum levels of xanthine oxidase (XO),
increases DPP-4 serum activity, and activates vascular cells (detected by expression of
endothelial adhesion molecules and inducible cyclooxygenase (Cox-20). This leads to the
infiltration of WBC to the vascular wall (detected by aortic FACS analysis for
myelomonocytic cells, inducible nitric oxide synthase (Nos-2), Nox2, and myeloperoxidase
(MPO) expression) and oxidative damage of the vasculature (Nox1 expression, ROS
formation, 3-nitrotyrosine levels, and lipid peroxidation by malondialdehyde (MDA)).
Finally, the tissue damage results in smooth muscle constriction and endothelial
dysfunction. The green lines on the arrows define the inhibitory effects of linagliptin on
septic complications. Adapted from [208]. With permission by Oxford University Press.
Copyright 2012, Oxford University Press.
These promising results encouraged us to investigate the impact of DPP-4 inhibition and GLP-1
supplementation on the survival of endotoxemic mice [207]. Pre- as well as post-treatment with linagliptin and the GLP-1 analog liraglutide improved survival of endotoxemic animals significantly (Figure 3A). As a proof of concept, we tested the survival of LPS-treated DPP-4−/− mice and found that
Int. J. Mol. Sci. 2015, 16
these mice are also protected from endotoxic shock-dependent death [207]. Ku et al. found similar results in DPP-4−/− rats and postulated that increased GLP-1 levels are responsible for the improved survival [244]. In line with these murine data, the oxidative burst in whole blood of LPS-treated rats was significantly increased and normalized by DPP-4 inhibition and GLP-1 supplementation (Figure 3B). In accordance with this observation, the nitrosyl-iron hemoglobin (Hb-NO) signal was measured by electron spin resonance (EPR) spectroscopy in whole blood as a direct read-out for increased iNOS activity in LPS-treated animals, which was increased in endotoxemic rats and normalized by linagliptin and liraglutide therapy (Figure 3C). As a marker of vascular oxidative stress, the dihydoethidium fluorescence signal was increased in the vascular wall of LPS-treated rats and normalized by linagliptin and liraglutide treatment (Figure 3D). In summary, these data nicely show the anti-inflammatory and antioxidant potential of DPP-4 inhibitors and GLP-1 analogs, but also reveal substantial differences between the different DPP-4 inhibiting drugs that might be related to their binding affinity and specific location in the active site of DPP-4 [207,208].
A known adverse side effect of DPP-4 inhibition in humans is an increased risk for infections like
nasopharygitis (risk ratio, 1.2 (95% CI, 1.0–1.4)) or urinary tract infection (risk ratio 1.5 (95% CI, 1.0–2.2)). These results from a meta-analysis reflect the abovementioned immunomodulatory effects of DPP-4 inhibition [245]. Furthermore, the GLP-1 analog exendin-4 reduced inflammation in a non-alcoholic steatohepatitis (NASH) animal model by decreasing the infiltration of macrophages (CD68+, F4/80+) [246]. Sitagliptin improved inflammation and fibrosis in methionine/choline-deficient diet-induced steatohepatitis [247]. NASH is a liver inflammatory disease, sharing several features with atherosclerosis [248]. Besides atherosclerosis, sepsis, and NASH, DPP-4 inhibitors and GLP-1 analogs exert immunomodulatory effects in various models of chronic inflammatory diseases such as colitis [249], asthma [250], chronic obstructive lung disease (COPD) [251], and rheumatoid arthritis [252].
Figure 3. Cont.
Int. J. Mol. Sci. 2015, 16
Figure 3. Protective effects of dipeptidyl peptidase-4 inhibition and glucagon-like
peptide-1 analog supplementation in an animal model of LPS-induced endotoxemia.
(A) Survival of animals was recorded and interpreted by Kaplan–Meier curves; (B) Whole
blood oxidative burst upon stimulation with the fungal endotoxin zymosan A was measured
by chemiluminescence using the luminol analog L-012; (C) iNOS-derived nitric oxide
was determined in whole blood by measurement of nitrosyl-iron hemoglobin by electron
paramagnetic resonance spectroscopy; (D) Vascular ROS formation was measured in aortic
cryo-sections by dehydrothidium (DHE)-dependent oxidative fluorescence microtopography.
Data are mean ± SEM of experiments with 19–36 mice (A) or at least three rats per group
(B–D). * p < 0.05 vs. Ctr; # p < 0.05 vs. LPS. Adapted from [207]. With permission by
Springer-Verlag Berlin Heidelberg. Copyright 2015, Springer.
4. Immunomodulation as a Therapeutic Strategy
As already outlined above, inflammation represents an independent risk factor for the development of
cardiovascular disease. C-reactive protein (CRP) is an acute phase protein and indicates inflammatory processes. According to the PROVE IT-TIMI 22 trial of patients with acute coronary syndrome after initiation of statin therapy, the risk of recurrent myocardial infarction or coronary death was significantly elevated in patients with a high hsCPR (>2 mg/L) compared to patients with low hsCPR levels [253]. The cytokine IL-17 was demonstrated to induce death of human endothelial cells, contributing to plaque destabilization and acute coronary syndrome by disruption of the blood-brain-barrier
Int. J. Mol. Sci. 2015, 16
and activation of NADPH oxidase in brain endothelial cells [254,255]. These adverse effects were suppressed by administration of an IL-17A blocking antibody or by antioxidant therapy [255]. IL-17 induces endothelial cell activation, expression of endothelial adhesion molecules, followed by adhesion and infiltration of neutrophils [256], providing the rational for beneficial effects of therapy with a soluble TNF-α receptor antibody (etanercept) on angiotensin-II induced vascular superoxide production and hypertension [257,258]. The immunosuppressive drug methotrexate (MTX) is used in treatment of cancer and autoimmune disease. Rheumatoid arthritis patients suffer from chronic inflammation and have an increased risk for cardiovascular events. A meta-analysis revealed the antirheumatic drug to be protective against cardiovascular disease in patients with chronic inflammation [259]. Studies on myocardial infarction in dogs showed reduced infarction size by methotrexate treatment [260]. Based on these interesting results, the ongoing TETHYS trial, which investigates the effects of MTX therapy on myocardial infarction with ST-segment elevation, was initiated [261]. An additional clinical trial, which faces the effect of immunosuppression by MTX on the cardiovascular system, is the ongoing CIRT trial (Cardiovascular Inflammation Reduction trial). Patients with myocardial infarction and either type 2 diabetes mellitus or metabolic syndrome will be treated with low-dose MTX or a placebo [262]. Another target currently under investigation for immunomodulation in cardiovascular disease is IL-1β. Animal studies revealed that blockade of IL-1β improves endothelial regrowth, reduces neointima formation, and thereby prevents restenosis following carotid denudation [263]. Furthermore, it ameliorates cardiac remodeling and reduces cardiomyocyte apoptosis after experimental acute myocardial infarction [264]. The ongoing CANTOS trial examines the cardiovascular outcome after blockade of IL-1 β by canakinumab in post myocardial infarction patients [265]. There is convincing evidence for a contribution of inflammation to cardiovascular disease and immunomodulation is a promising new therapeutic approach to treat cardiovascular disease. Nevertheless, it is challenging to find a specific target and tool for modulating the inflammatory cascade. The ongoing clinical trials will provide more answers for these questions.
5. Conclusions
Endothelial dysfunction is an early hallmark of most cardiovascular disease in general [266] and
coronary heart disease in particular [13], as well as related future cardiovascular events. Oxidative stress is associated with cardiovascular disease [1,2] but also represents a prognostic marker of future cardiovascular events [12]. Therefore, oxidative stress must be considered a trigger of cardiovascular events; it probably contributes to the progression of cardiovascular disease and represents an attractive target for its therapy [267]. According to more recent data, there is a close correlation between oxidative stress and inflammation in the vasculature [8,28,218,268], making both of them independent triggers/risk factors for the progression of cardiovascular disease and future cardiovascular events [12,36]. Since large clinical trials on chronic oral, systemic, unspecific antioxidant therapy failed to display beneficial effects on cardiovascular events [269], the use of source and cell (organelle)-specific compounds or activators of intrinsic antioxidant systems represents a more promising strategy [122]. Another attractive attempt might be the exploitation of the antioxidant and anti-inflammatory properties of established cardiovascular drugs [5]. Screening for candidates with potent anti-inflammatory effects could represent important additional criteria for the development of
Int. J. Mol. Sci. 2015, 16
cardiovascular drugs in the future. Comparison of drugs with similar primary effects (e.g., blood pressure lowering) but with or without pleiotropic anti-inflammatory and antioxidant effects will allow us to study the importance of these pleiotropic effects. Also, prescreening of patients for markers of inflammation and/or oxidative stress will help us to develop or find the most efficient drug or drug combination for the treatment of the patients in an individual way (in the sense of personalized medicine).
The results obtained with GLP-1 supplementation and DPP-4 inhibition in models of endotoxemia
are quite promising, but they remind us of the enthusiasm about statins as a new treatment strategy in sepsis and their failure in large clinical trials [270]. Animal trials and small clinical trials showed convincing evidence for a mortality reduction by statin therapy, which also relied on immunomodulatory effects [271]. Unfortunately, these results could not be reproduced in a large multi-center trial [243] and meta-analysis revealed no improvement of survival [272]. More research is needed to better characterize the antioxidant and anti-inflammatory effects of DPP-4 inhibition and GLP-1 supplementation in the pathogenesis of sepsis. Studies in different models of sepsis (i.e., acute respiratory response syndrome or cecal ligation and puncture) and small clinical trials could shed light on this promising field of sepsis research in the future.
We thank Thilo Weckmüller and Margot Neuser for expert graphical assistance. The present work
was supported by vascular biology research grants from Boehringer Ingelheim Pharma GmbH & Co. KG, Mainzer Herz Stiftung, and the Center for Translational Vascular Biology (CTVB) (to Andreas Daiber and Thomas Münzel). Sebastian Steven holds a Virchow Fellowship from the Center of Thrombosis and Hemostasis (Mainz, Germany), funded by the Federal Ministry of Education and Research (BMBF 01EO1003). Andreas Daiber and Sebastian Steven were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU-ROS).
Author Contributions
All authors contributed equally to this work. Sebastian Steven and Andreas Daiber drafted the
manuscript. Thomas Münzel and Andreas Daiber made critical revisions.
Conflicts of Interest
The authors declare no conflict of interest.
References
Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic
mechanisms and in vivo monitoring of ROS. Circulation 2003, 108, 1912–1916.
Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: Animal
and human studies. Circulation 2003, 108, 2034–2040.
Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.
Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications.
Ann. Med. 2011, 43, 259–272.
Int. J. Mol. Sci. 2015, 16
Chen, A.F.; Chen, D.D.; Daiber, A.; Faraci, F.M.; Li, H.; Rembold, C.M.; Laher, I. Free radical
biology of the cardiovascular system. Clin. Sci. 2012, 123, 73–91.
Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of
mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal.
2014, 20, 308–324.
Daiber, A.; Oelze, M.; Daub, S.; Steven, S.; Schuff, A.; Kroller-Schon, S.; Hausding, M.; Wenzel, P.; Schulz, E.; Gori, T.; et al. Vascular redox signaling, redox switches in endothelial nitric oxide synthase and endothelial dysfunction. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer-Verlag: Berlin Heidelberg, Germany, 2014; pp. 1177–1211.
Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A.E. eNOS uncoupling in
cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014,
20, 3579–3594.
Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.;
Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140.
10. Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel
to menace. Circulation 2006, 113, 1708–1714.
11. Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric
oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the
cgmp-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557.
12. Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative
stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation
2001, 104, 2673–2678.
13. Schachinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator
dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101,
1899–1906.
14. Lin, C.P.; Lin, F.Y.; Huang, P.H.; Chen, Y.L.; Chen, W.C.; Chen, H.Y.; Huang, Y.C.;
Liao, W.L.; Huang, H.C.; Liu, P.L.; et al. Endothelial progenitor cell dysfunction in
cardiovascular diseases: Role of reactive oxygen species and inflammation. BioMed Res. Int.
2013, 2013, 845037.
15. Assmus, B.; Leistner, D.M.; Schachinger, V.; Erbs, S.; Elsasser, A.; Haberbosch, W.;
Hambrecht, R.; Sedding, D.; Yu, J.; Corti, R.; et al. Long-term clinical outcome after
intracoronary application of bone marrow-derived mononuclear cells for acute myocardial
infarction: Migratory capacity of administered cells determines event-free survival. Eur. Heart J.
2014, 35, 1275–1283.
16. Schachinger, V.; Erbs, S.; Elsasser, A.; Haberbosch, W.; Hambrecht, R.; Holschermann, H.;
Yu, J.; Corti, R.; Mathey, D.G.; Hamm, C.W.; et al. Intracoronary bone marrow-derived
progenitor cells in acute myocardial infarction. N. Engl. J. Med. 2006, 355, 1210–1221.
17. Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide
anion production. J. Clin. Investig. 1993, 91, 2546–2551.
18. Harrison, D.G.; Ohara, Y. Physiologic consequences of increased vascular oxidant stresses in
hypercholesterolemia and atherosclerosis: Implications for impaired vasomotion. Am. J. Cardiol.
1995, 75, 75B–81B.
Int. J. Mol. Sci. 2015, 16
19. Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores
and reactive oxygen species. Biochim. Biophys. Acta. 2010, 1797, 897–906.
20. Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and
ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950.
21. Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M.
NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8,
691–728.
22. Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.;
Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced
hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460.
23. Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.;
Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin
II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381.
24. Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.;
Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of
proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS).
J. Exp. Med. 2011, 208, 519–533.
25. West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.;
Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. Tlr signalling augments macrophage bactericidal
activity through mitochondrial ROS. Nature 2011, 472, 476–480.
26. Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome
activation. Nature 2011, 469, 221–225.
27. Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in
inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167.
28. El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in
aging. Free Radic. Biol. Med. 2013, 65, 380–401.
29. Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial oxidative stress, mitochondrial DNA damage
and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953.
30. Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.;
Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between
mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells
and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266.
31. Herrera, J.; Ferrebuz, A.; MacGregor, E.G.; Rodriguez-Iturbe, B. Mycophenolate mofetil treatment
improves hypertension in patients with psoriasis and rheumatoid arthritis. J. Am. Soc. Nephrol.
2006, 17, S218–S225.
32. Soltesz, P.; Kerekes, G.; Der, H.; Szucs, G.; Szanto, S.; Kiss, E.; Bodolay, E.; Zeher, M.; Timar, O.;
Szodoray, P.; et al. Comparative assessment of vascular function in autoimmune rheumatic
diseases: Considerations of prevention and treatment. Autoimmun. Rev. 2011, 10, 416–425.
33. Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spano, F.; Puppo, F. Endothelial
dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317.
34. Vena, G.A.; Vestita, M.; Cassano, N. Psoriasis and cardiovascular disease. Dermatol. Ther. 2010,
23, 144–151.
Int. J. Mol. Sci. 2015, 16
35. Hak, A.E.; Karlson, E.W.; Feskanich, D.; Stampfer, M.J.; Costenbader, K.H. Systemic lupus
erythematosus and the risk of cardiovascular disease: Results from the nurses' health study.
Arthritis Rheum. 2009, 61, 1396–1402.
36. Mehta, N.N.; Azfar, R.S.; Shin, D.B.; Neimann, A.L.; Troxel, A.B.; Gelfand, J.M. Patients with
severe psoriasis are at increased risk of cardiovascular mortality: Cohort study using the general
practice research database. Eur. Heart J. 2010, 31, 1000–1006.
37. Peters, M.J.; Symmons, D.P.; McCarey, D.; Dijkmans, B.A.; Nicola, P.; Kvien, T.K.;
McInnes, I.B.; Haentzschel, H.; Gonzalez-Gay, M.A.; Provan, S.; et al. Eular evidence-based
recommendations for cardiovascular risk management in patients with rheumatoid arthritis and
other forms of inflammatory arthritis. Ann. Rheum. Dis. 2010, 69, 325–331.
38. Sodergren, A.; Karp, K.; Boman, K.; Eriksson, C.; Lundstrom, E.; Smedby, T.; Soderlund, L.;
Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Atherosclerosis in early rheumatoid arthritis: Very
early endothelial activation and rapid progression of intima media thickness. Arthritis Res. Ther.
2010, 12, R158.
39. Balci, D.D.; Balci, A.; Karazincir, S.; Ucar, E.; Iyigun, U.; Yalcin, F.; Seyfeli, E.; Inandi, T.;
Egilmez, E. Increased carotid artery intima-media thickness and impaired endothelial function in
psoriasis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1–6.
40. Di Cesare, A.; di Meglio, P.; Nestle, F.O. The IL-23/Th17 axis in the immunopathogenesis of
psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350.
41. Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.;
Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis.
N. Engl. J. Med. 2012, 366, 1190–1199.
42. Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.;
Russell, C.B.; Thompson, E.H.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for
psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189.
43. Crispin, J.C.; Tsokos, G.C. IL-17 in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010,
2010, 943254.
44. Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid
arthritis. Rheumatology (Oxford) 2012, 51 (Suppl. 5), v3–v11.
45. Pasceri, V.; Yeh, E.T. A tale of two diseases: Atherosclerosis and rheumatoid arthritis.
Circulation 1999, 100, 2124–2126.
46. Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.;
Glynn, R.J.; Buring, J.E.; Gaziano, J.M. Multivitamins in the prevention of cardiovascular
disease in men: The physicians' health study II randomized controlled trial. JAMA 2012, 308,
1751–1760.
47. Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.; Ross, C.; Arnold, A.;
Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin e supplementation on
cardiovascular events and cancer: A randomized controlled trial. JAMA 2005, 293, 1338–1347.
48. Muntwyler, J.; Hennekens, C.H.; Manson, J.E.; Buring, J.E.; Gaziano, J.M. Vitamin supplement
use in a low-risk population of us male physicians and subsequent cardiovascular mortality.
Arch. Intern. Med. 2002, 162, 1472–1476.
Int. J. Mol. Sci. 2015, 16
49. Mann, J.F.; Lonn, E.M.; Yi, Q.; Gerstein, H.C.; Hoogwerf, B.J.; Pogue, J.; Bosch, J.;
Dagenais, G.R.; Yusuf, S. Effects of vitamin e on cardiovascular outcomes in people with
mild-to-moderate renal insufficiency: Results of the hope study. Kidney Int. 2004, 65,
1375–1380.
50. Yusuf, S.; Dagenais, G.; Pogue, J.; Bosch, J.; Sleight, P. Vitamin e supplementation and
cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study
investigators. N. Engl. J. Med. 2000, 342, 154–160.
51. Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.P.; Daiber, A.; Cuadrado, A.
Antioxidants in translational medicine. Antioxid. Redox Signal. 2015, doi:10.1089/ars.2015.6393.
52. Lee, D.H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R., Jr. Does supplemental
vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004,
80, 1194–1200.
53. Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.;
Ashwood, T.; Wasiewski, W.W.; Emeribe, U.; et al. Nxy-059 for the treatment of acute ischemic
stroke. N. Engl. J. Med. 2007, 357, 562–571.
54. Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized
trials of antioxidant supplements for primary and secondary prevention: Systematic review and
meta-analysis. JAMA 2007, 297, 842–857.
55. Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention
of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228.
56. Harris, H.R.; Orsini, N.; Wolk, A. Vitamin C and survival among women with breast cancer:
A meta-analysis. Eur. J. Cancer 2014, 50, 1223–1231.
57. Ashor, A.W.; Lara, J.; Mathers, J.C.; Siervo, M. Effect of vitamin C on endothelial function in
health and disease: A systematic review and meta-analysis of randomised controlled trials.
Atherosclerosis 2014, 235, 9–20.
58. Heitzer, T.; Finckh, B.; Albers, S.; Krohn, K.; Kohlschutter, A.; Meinertz, T. Beneficial effects of
α-lipoic acid and ascorbic acid on endothelium-dependent, nitric oxide-mediated vasodilation in
diabetic patients: Relation to parameters of oxidative stress. Free Radic. Biol. Med. 2001, 31,
53–61.
59. Heitzer, T.; Brockhoff, C.; Mayer, B.; Warnholtz, A.; Mollnau, H.; Henne, S.; Meinertz, T.;
Munzel, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic
smokers : Evidence for a dysfunctional nitric oxide synthase. Circ. Res. 2000, 86, E36–E41.
60. Heitzer, T.; Just, H.; Munzel, T. Antioxidant vitamin C improves endothelial dysfunction in
chronic smokers. Circulation 1996, 94, 6–9.
61. Levine, M.; Rumsey, S.C.; Daruwala, R.; Park, J.B.; Wang, Y. Criteria and recommendations for
vitamin C intake. JAMA 1999, 281, 1415–1423.
62. Deng, Y.B.; Li, T.L.; Xiang, H.J.; Chang, Q.; Li, C.L. Impaired endothelial function in the
brachial artery after kawasaki disease and the effects of intravenous administration of vitamin C.
Pediatr. Infect. Dis. J. 2003, 22, 34–39.
63. Schaufele, T.G.; Schlaich, M.P.; Delles, C.; Klingbeil, A.U.; Fleischmann, E.H.; Schmieder, R.E.
Impaired basal no activity in patients with glomerular disease and the influence of oxidative
stress. Kidney Int. 2006, 70, 1177–1181.
Int. J. Mol. Sci. 2015, 16
64. Solzbach, U.; Hornig, B.; Jeserich, M.; Just, H. Vitamin C improves endothelial dysfunction of
epicardial coronary arteries in hypertensive patients. Circulation 1997, 96, 1513–1519.
65. Hernandez-Guerra, M.; Garcia-Pagan, J.C.; Turnes, J.; Bellot, P.; Deulofeu, R.; Abraldes, J.G.;
Bosch, J. Ascorbic acid improves the intrahepatic endothelial dysfunction of patients with
cirrhosis and portal hypertension. Hepatology 2006, 43, 485–491.
66. Hagel, A.F.; Layritz, C.M.; Hagel, W.H.; Hagel, H.J.; Hagel, E.; Dauth, W.; Kressel, J.; Regnet, T.;
Rosenberg, A.; Neurath, M.F.; et al. Intravenous infusion of ascorbic acid decreases serum
histamine concentrations in patients with allergic and non-allergic diseases. Naunyn Schmiedebergs
Arch. Pharmacol. 2013, 386, 789–793.
67. Kang, H.S.; Park, J.J.; Ahn, S.K.; Hur, D.G.; Kim, H.Y. Effect of high dose intravenous
vitamin C on idiopathic sudden sensorineural hearing loss: A prospective single-blind
randomized controlled trial. Eur. Arch. Otorhinolaryngol. 2013, 270, 2631–2636.
68. Du, W.D.; Yuan, Z.R.; Sun, J.; Tang, J.X.; Cheng, A.Q.; Shen, D.M.; Huang, C.J.; Song, X.H.;
Yu, X.F.; Zheng, S.B. Therapeutic efficacy of high-dose vitamin C on acute pancreatitis and its
potential mechanisms. World J. Gastroenterol. 2003, 9, 2565–2569.
69. Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281.
70. Harris, R.A.; Pedersen-White, J.; Guo, D.H.; Stallmann-Jorgensen, I.S.; Keeton, D.; Huang, Y.;
Shah, Y.; Zhu, H.; Dong, Y. Vitamin D3 supplementation for 16 weeks improves flow-mediated
dilation in overweight african-american adults. Am. J. Hypertens. 2011, 24, 557–562.
71. Sugden, J.A.; Davies, J.I.; Witham, M.D.; Morris, A.D.; Struthers, A.D. Vitamin D improves
endothelial function in patients with type 2 diabetes mellitus and low vitamin D levels. Diabet. Med.
2008, 25, 320–325.
72. Pfeifer, M.; Begerow, B.; Minne, H.W.; Nachtigall, D.; Hansen, C. Effects of a short-term
vitamin D3 and calcium supplementation on blood pressure and parathyroid hormone levels in
elderly women. J. Clin. Endocrinol. Metab. 2001, 86, 1633–1637.
73. Kim, H.W.; Park, C.W.; Shin, Y.S.; Kim, Y.S.; Shin, S.J.; Kim, Y.S.; Choi, E.J.; Chang, Y.S.;
Bang, B.K. Calcitriol regresses cardiac hypertrophy and QT dispersion in secondary
hyperparathyroidism on hemodialysis. Nephron. Clin. Pract. 2006, 102, c21–29.
74. Park, C.W.; Oh, Y.S.; Shin, Y.S.; Kim, C.M.; Kim, Y.S.; Kim, S.Y.; Choi, E.J.; Chang, Y.S.;
Bang, B.K. Intravenous calcitriol regresses myocardial hypertrophy in hemodialysis patients with
secondary hyperparathyroidism. Am. J. Kidney Dis. 1999, 33, 73–81.
75. Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Wetterslev, J.; Simonetti, R.G.;
Bjelakovic, M.; Gluud, C. Vitamin D supplementation for prevention of mortality in adults.
Cochrane Database Syst. Rev. 2014, 1, CD007470.
76. Janssen, H.C.; Samson, M.M.; Verhaar, H.J. Vitamin D deficiency, muscle function, and falls in
elderly people. Am. J. Clin. Nutr. 2002, 75, 611–615.
77. Mozos, I.; Marginean, O. Links between vitamin D deficiency and cardiovascular diseases.
BioMed Res. Int. 2015, 2015, 109275.
78. Stocker, R. The ambivalence of vitamin E in atherogenesis. Trends Biochem. Sci. 1999, 24,
Int. J. Mol. Sci. 2015, 16
79. Khaw, K.T.; Bingham, S.; Welch, A.; Luben, R.; Wareham, N.; Oakes, S.; Day, N. Relation
between plasma ascorbic acid and mortality in men and women in epic-norfolk prospective
study: A prospective population study. European prospective investigation into cancer and
nutrition. Lancet 2001, 357, 657–663.
80. Chen, G.C.; Lu, D.B.; Pang, Z.; Liu, Q.F. Vitamin C intake, circulating vitamin C and risk of
stroke: A meta-analysis of prospective studies. J. Am. Heart Assoc. 2013, 2, e000329.
81. Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle-associated vascular
disease progression? Evidence based on experimental and clinical studies. Antioxid. Redox Signal.
2013, 19, 2084–2104.
82. Ashor, A.W.; Siervo, M.; Lara, J.; Oggioni, C.; Mathers, J.C. Antioxidant vitamin
supplementation reduces arterial stiffness in adults: A systematic review and meta-analysis of
randomized controlled trials. J. Nutr. 2014, 144, 1594–1602.
83. Koopman, W.J.; Verkaart, S.; Visch, H.J.; van der Westhuizen, F.H.; Murphy, M.P.;
van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Inhibition of complex I of the electron transport chain causes O −
2 ·-mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol.
2005, 288, C1440–C1450.
84. Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.;
Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion
injury. FASEB J. 2005, 19, 1088–1095.
85. Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.;
Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction
drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive
pulmonary disease. J. Allergy Clin. Immunol. 2015, doi:10.1016/j.jaci.2015.01.046.
86. Esplugues, J.V.; Rocha, M.; Nunez, C.; Bosca, I.; Ibiza, S.; Herance, J.R.; Ortega, A.;
Serrador, J.M.; D'Ocon, P.; Victor, V.M. Complex i dysfunction and tolerance to nitroglycerin:
An approach based on mitochondrial-targeted antioxidants. Circ. Res. 2006, 99, 1067–1075.
87. Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cocheme, H.M.;
Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves
endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328.
88. Miquel, E.; Cassina, A.; Martinez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodriguez-Bottero, S.;
Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the
mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis.
Free Radic. Biol. Med. 2014, 70, 204–213.
89. Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.;
Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates
experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory
cytokines. BMC Med. 2013, 11, 178.
90. Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.;
Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in
Ins2(+/)−(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432,
9–19.
Int. J. Mol. Sci. 2015, 16
91. Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.;
Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension.
Circ. Res. 2010, 107, 106–116.
92. Batinic-Haberle, I.; Rajic, Z.; Tovmasyan, A.; Reboucas, J.S.; Ye, X.; Leong, K.W.;
Dewhirst, M.W.; Vujaskovic, Z.; Benov, L.; Spasojevic, I. Diverse functions of cationic Mn(III)
N-substituted pyridylporphyrins, recognized as SOD mimics. Free Radic. Biol. Med. 2011, 51,
1035–1053.
93. Batinic-Haberle, I.; Tovmasyan, A.; Roberts, E.R.; Vujaskovic, Z.; Leong, K.W.; Spasojevic, I.
SOD therapeutics: Latest insights into their structure-activity relationships and impact on the
cellular redox-based signaling pathways. Antioxid. Redox Signal. 2014, 20, 2372–2415.
94. Smith, R.A.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics
and probes. Antioxid. Redox Signal. 2011, 15, 3021–3038.
95. Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med.
2014, 66, 20–23.
96. Shuvaev, V.V.; Christofidou-Solomidou, M.; Bhora, F.; Laude, K.; Cai, H.; Dikalov, S.;
Arguiri, E.; Solomides, C.C.; Albelda, S.M.; Harrison, D.G.; et al. Targeted detoxification of
selected reactive oxygen species in the vascular endothelium. J. Pharmacol. Exp. Ther. 2009, 331,
404–411.
97. Fernandez, C.; Hattan, C.M.; Kerns, R.J. Semi-synthetic heparin derivatives: Chemical
modifications of heparin beyond chain length, sulfate substitution pattern and N-sulfo/N-acetyl
groups. Carbohydr. Res. 2006, 341, 1253–1265.
98. Kleschyov, A.L.; Sen, V.; Golubev, V.; Munnemann, K.; Hinderberger, D.; Lackner, K.J.;
Weber, S.; Terekhov, M.; Schreiber, L.M.; Munzel, T. Heparin-polynitroxides: Synthesis and
preliminary evaluation as cardiovascular EPR/MR imaging probes and extracellular space-targeted
antioxidants. Eur. J. Med. Chem. 2012, 58, 265–271.
99. Kleschyov, A.L.; Sen, V.D. Heparin-polynitroxide derivatives: A platform for new diagnostic
and therapeutic agents in cardiovascular disease? Future Med. Chem. 2013, 5, 385–388.
100. Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by
effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5,
275–289.
101. Hayes, P.; Knaus, U.G. Balancing reactive oxygen species in the epigenome: NADPH oxidases
as target and perpetrator. Antioxid. Redox Signal. 2013, 18, 1937–1945.
102. Baird, A.M.; O'Byrne, K.J.; Gray, S.G. Reactive oxygen species and reactive nitrogen species in
epigenetic modifications. In Systems Biology of Free Radicals and Antioxidants; Laher, I. Ed.; Springer-Verlag: Berlin Heidelberg, Germany, 2014; pp. 437–455.
103. Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.;
Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of
mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive
cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671.
104. Cooper, M.P.; Keaney, J.F., Jr. Epigenetic control of angiogenesis via DNA methylation.
Circulation 2011, 123, 2916–2918.
Int. J. Mol. Sci. 2015, 16
105. Kim, G.H.; Ryan, J.J.; Archer, S.L. The role of redox signaling in epigenetics and cardiovascular
disease. Antioxid. Redox Signal. 2013, 18, 1920–1936.
106. Wang, J.; Gong, L.; Tan, Y.; Hui, R.; Wang, Y. Hypertensive epigenetics: From DNA
methylation to micrornas. J. Hum. Hypertens. 2015, doi:10.1038/jhh.2014.132.
107. Li, H.; Xia, N.; Forstermann, U. Cardiovascular effects and molecular targets of resveratrol.
Nitric Oxide 2012, 26, 102–110.
108. Mukhopadhyay, P.; Pacher, P.; Das, D.K. Microrna signatures of resveratrol in the ischemic
heart. Ann. N. Y. Acad. Sci. 2011, 1215, 109–116.
109. Farghali, H.; Kutinova Canova, N.; Lekic, N. Resveratrol and related compounds as antioxidants
with an allosteric mechanism of action in epigenetic drug targets. Physiol. Res. 2013, 62, 1–13.
110. Gatz, S.A.; Wiesmuller, L. Take a break—resveratrol in action on DNA. Carcinogenesis 2008,
29, 321–332.
111. Zhou, W.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Clinical use and
molecular mechanisms of action of extract of ginkgo biloba leaves in cardiovascular diseases.
Cardiovasc. Drug Rev. 2004, 22, 309–319.
112. Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhofer, S.; Radermacher, K.A.;
Janssen, B.; Gorlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and
molecular target in ischemia/reperfusion injury. Int. J. Mol. Med. 2012, 90, 1391–1406.
113. Altenhofer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.;
Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The Nox toolbox: Validating the role of
NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 2012, 69, 2327–2343.
114. Radermacher, K.A.; Wingler, K.; Langhauser, F.; Altenhofer, S.; Kleikers, P.; Hermans, J.J.;
Hrabe de Angelis, M.; Kleinschnitz, C.; Schmidt, H.H. Neuroprotection after stroke by targeting
Nox4 as a source of oxidative stress. Antioxid. Redox Signal. 2013, 18, 1418–1427.
115. Wingler, K.; Hermans, J.J.; Schiffers, P.; Moens, A.; Paul, M.; Schmidt, H.H. Nox1, 2, 4, 5:
Counting out oxidative stress. Br. J. Pharmacol. 2011, 164, 866–883.
116. Lee, B.E.; Toledo, A.H.; Anaya-Prado, R.; Roach, R.R.; Toledo-Pereyra, L.H. Allopurinol,
xanthine oxidase, and cardiac ischemia. J. Investig. Med. 2009, 57, 902–909.
117. Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase
inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis.
Cardiovasc. Ther. 2012, 30, 217–226.
118. Jazwa, A.; Cuadrado, A. Targeting heme oxygenase-1 for neuroprotection and
neuroinflammation in neurodegenerative diseases. Curr. Drug Targets 2010, 11, 1517–1531.
119. Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P.
NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and
therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768.
120. Follmann, M.; Griebenow, N.; Hahn, M.G.; Hartung, I.; Mais, F.J.; Mittendorf, J.; Schafer, M.;
Schirok, H.; Stasch, J.P.; Stoll, F.; et al. The chemistry and biology of soluble guanylate cyclase
stimulators and activators. Angew. Chem. Int. Ed. Engl. 2013, 52, 9442–9462.
121. Schmidt, H.H.; Schmidt, P.M.; Stasch, J.P. NO- and haem-independent soluble guanylate cyclase
activators. Handb. Exp. Pharmacol. 2009, 191, 309–339.
Int. J. Mol. Sci. 2015, 16
122. Lonn, M.E.; Dennis, J.M.; Stocker, R. Actions of "antioxidants" in the protection against
atherosclerosis. Free Radic. Biol. Med. 2012, 53, 863–884.
123. Drexler, H.; Hornig, B. Endothelial dysfunction in human disease. J. Mol. Cell. Cardiol. 1999,
31, 51–60.
124. Warnholtz, A.; Munzel, T. Why do antioxidants fail to provide clinical benefit? Curr. Control.
Trials Cardiovasc. Med. 2000, 1, 38–40.
125. Porsti, I.; Bara, A.T.; Busse, R.; Hecker, M. Release of nitric oxide by angiotensin-(1–7) from
porcine coronary endothelium: Implications for a novel angiotensin receptor. Br. J. Pharmacol.
1994, 111, 652–654.
126. Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.;
Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of increased vascular superoxide
production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler. Thromb.
Vasc. Biol. 2005, 25, 2554–2559.
127. Williams, H.C.; Griendling, K.K. NADPH oxidase inhibitors: New antihypertensive agents?
J. Cardiovasc. Pharmacol. 2007, 50, 9–16.
128. Soehnlein, O.; Schmeisser, A.; Cicha, I.; Reiss, C.; Ulbrich, H.; Lindbom, L.; Daniel, W.G.;
Garlichs, C.D. Ace inhibition lowers angiotensin-II-induced monocyte adhesion to HUVEC by
reduction of p65 translocation and AT1 expression. J. Vasc. Res. 2005, 42, 399–407.
129. Caspritz, G.; Alpermann, H.G.; Schleyerbach, R. Influence of the new angiotensin converting
enzyme inhibitor ramipril on several models of acute inflammation and the adjuvant arthritis in
the rat. Arzneimittelforschung 1986, 36, 1605–1608.
130. Durik, M.; Seva Pessoa, B.; Roks, A.J. The renin-angiotensin system, bone marrow and
progenitor cells. Clin. Sci. 2012, 123, 205–223.
131. Munzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative
stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart. J. 2015,
doi:10.1093/eurheartj/ehv305.
132. Takemoto, M.; Liao, J.K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a
reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1712–1719.
133. Ray, K.K.; Cannon, C.P. Pathological changes in acute coronary syndromes: The role of
statin therapy in the modulation of inflammation, endothelial function and coagulation.
J. Thromb. Thrombolysis 2004, 18, 89–101.
134. Patel, T.N.; Shishehbor, M.H.; Bhatt, D.L. A review of high-dose statin therapy: Targeting
cholesterol and inflammation in atherosclerosis. Eur. Heart J. 2007, 28, 664–672.
135. Adam, O.; Laufs, U. Rac1-mediated effects of HMG-CoA reductase inhibitors (statins) in
cardiovascular disease. Antioxid. Redox Signal. 2014, 20, 1238–1250.
136. Wenzel, P.; Daiber, A.; Oelze, M.; Brandt, M.; Closs, E.; Xu, J.; Thum, T.; Bauersachs, J.;
Ertl, G.; Zou, M.H.; et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase
inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis 2008, 198,
65–76.
137. Margaritis, M.; Channon, K.M.; Antoniades, C. Statins as regulators of redox state in the
vascular endothelium: Beyond lipid lowering. Antioxid. Redox Signal. 2014, 20, 1198–1215.
Int. J. Mol. Sci. 2015, 16
138. Habeos, I.G.; Ziros, P.G.; Chartoumpekis, D.; Psyrogiannis, A.; Kyriazopoulou, V.;
Papavassiliou, A.G. Simvastatin activates Keap1/Nrf2 signaling in rat liver. Int. J. Mol. Med.
2008, 86, 1279–1285.
139. Ali, F.; Zakkar, M.; Karu, K.; Lidington, E.A.; Hamdulay, S.S.; Boyle, J.J.; Zloh, M.; Bauer, A.;
Haskard, D.O.; Evans, P.C.; et al. Induction of the cytoprotective enzyme heme oxygenase-1 by
statins is enhanced in vascular endothelium exposed to laminar shear stress and impaired by
disturbed flow. J. Biol. Chem. 2009, 284, 18882–18892.
140. Hibbert, B.; Simard, T.; Ramirez, F.D.; Pourdjabbar, A.; Raizman, J.E.; Maze, R.; Wilson, K.R.;
Hawken, S.; O'Brien, E.R. The effect of statins on circulating endothelial progenitor cells in
humans: A systematic review. J. Cardiovasc. Pharmacol. 2013, 62, 491–496.
141. Magee, L.A.; Abalos, E.; von Dadelszen, P.; Sibai, B.; Easterling, T.; Walkinshaw, S.; Group, C.S.
How to manage hypertension in pregnancy effectively. Br. J. Clin. Pharmacol. 2011, 72, 394–401.
142. Anand, I.S.; Tam, S.W.; Rector, T.S.; Taylor, A.L.; Sabolinski, M.L.; Archambault, W.T.;
Adams, K.F.; Olukotun, A.Y.; Worcel, M.; Cohn, J.N. Influence of blood pressure on the
effectiveness of a fixed-dose combination of isosorbide dinitrate and hydralazine in the
african-american heart failure trial. J. Am. Coll. Cardiol. 2007, 49, 32–39.
143. Taylor, A.L.; Ziesche, S.; Yancy, C.W.; Carson, P.; Ferdinand, K.; Taylor, M.; Adams, K.;
Olukotun, A.Y.; Ofili, E.; Tam, S.W.; et al. Early and sustained benefit on event-free survival
and heart failure hospitalization from fixed-dose combination of isosorbide dinitrate/hydralazine:
Consistency across subgroups in the african-american heart failure trial. Circulation 2007, 115,
1747–1753.
144. Cohn, J.N.; Tam, S.W.; Anand, I.S.; Taylor, A.L.; Sabolinski, M.L.; Worcel, M. Isosorbide
dinitrate and hydralazine in a fixed-dose combination produces further regression of left
ventricular remodeling in a well-treated black population with heart failure: Results from
A-HeFT. J. Card. Fail. 2007, 13, 331–339.
145. Daiber, A.; Oelze, M.; Coldewey, M.; Kaiser, K.; Huth, C.; Schildknecht, S.; Bachschmid, M.;
Nazirisadeh, Y.; Ullrich, V.; Mulsch, A.; et al. Hydralazine is a powerful inhibitor of
peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients
with congestive heart failure. Biochem. Biophys. Res. Commun. 2005, 338, 1865–1874.
146. Daiber, A.; Mulsch, A.; Hink, U.; Mollnau, H.; Warnholtz, A.; Oelze, M.; Munzel, T. The
oxidative stress concept of nitrate tolerance and the antioxidant properties of hydralazine.
Am. J. Cardiol. 2005, 96, 25i–36i.
147. Munzel, T.; Kurz, S.; Rajagopalan, S.; Thoenes, M.; Berrington, W.R.; Thompson, J.A.;
Freeman, B.A.; Harrison, D.G. Hydralazine prevents nitroglycerin tolerance by inhibiting
activation of a membrane-bound nadh oxidase. A new action for an old drug. J. Clin. Investig.
1996, 98, 1465–1470.
148. Sekiya, M.; Sato, M.; Funada, J.; Ohtani, T.; Akutsu, H.; Watanabe, K. Effects of the long-term
administration of nicorandil on vascular endothelial function and the progression of
arteriosclerosis. J. Cardiovasc. Pharmacol. 2005, 46, 63–67.
149. Yang, X.F. Chemiluminescence investigation of carbon dioxide-enhanced oxidation of dihydralazine
sulfate by peroxynitrite and its application to pharmaceutical analysis. Anal. Chim. Acta 2008,
616, 190–195.
Int. J. Mol. Sci. 2015, 16
150. Knowles, H.J.; Tian, Y.M.; Mole, D.R.; Harris, A.L. Novel mechanism of action for hydralazine:
Induction of hypoxia-inducible factor-1α, vascular endothelial growth factor, and angiogenesis
by inhibition of prolyl hydroxylases. Circ. Res. 2004, 95, 162–169.
151. Aslam, S. Cardiovascular disease in dialysis patients: Do some antihypertensive drugs have
specific antioxidant effects or is it just blood pressure reduction? Does antioxidant treatment
reduce the risk for cardiovascular disease? Curr. Opin. Nephrol. Hypertens. 2008, 17, 99–105.
152. Hoenig, M.R.; Bianchi, C.; Sellke, F.W. Hypoxia inducible factor-1 α, endothelial progenitor
cells, monocytes, cardiovascular risk, wound healing, cobalt and hydralazine: A unifying
hypothesis. Curr. Drug Targets 2008, 9, 422–435.
153. Brehm, B.R.; Wolf, S.C.; Bertsch, D.; Klaussner, M.; Wesselborg, S.; Schuler, S.;
Schulze-Osthoff, K. Effects of nebivolol on proliferation and apoptosis of human coronary artery
smooth muscle and endothelial cells. Cardiovasc. Res. 2001, 49, 430–439.
154. Georgescu, A.; Pluteanu, F.; Flonta, M.L.; Badila, E.; Dorobantu, M.; Popov, D. The cellular
mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur. J. Pharmacol.
2005, 508, 159–166.
155. Broeders, M.A.; Doevendans, P.A.; Bekkers, B.C.; Bronsaer, R.; van Gorsel, E.; Heemskerk, J.W.;
Egbrink, M.G.; van Breda, E.; Reneman, R.S.; van Der Zee, R. Nebivolol: A third-generation
β-blocker that augments vascular nitric oxide release: Endothelial β2—adrenergic receptor—Mediated
nitric oxide production. Circulation 2000, 102, 677–684.
156. Tzemos, N.; Lim, P.O.; MacDonald, T.M. Nebivolol reverses endothelial dysfunction in essential
hypertension: A randomized, double-blind, crossover study. Circulation 2001, 104, 511–514.
157. Mollnau, H.; Schulz, E.; Daiber, A.; Baldus, S.; Oelze, M.; August, M.; Wendt, M.; Walter, U.;
Geiger, C.; Agrawal, R.; et al. Nebivolol prevents vascular nos III uncoupling in experimental
hyperlipidemia and inhibits NADPH oxidase activity in inflammatory cells. Arterioscler. Thromb.
Vasc. Biol. 2003, 23, 615–621.
158. Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.;
Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide
formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats.
Hypertension 2006, 48, 677–684.
159. Pasini, A.F.; Garbin, U.; Nava, M.C.; Stranieri, C.; Davoli, A.; Sawamura, T.; Cascio, V.L.;
Cominacini, L. Nebivolol decreases oxidative stress in essential hypertensive patients and
increases nitric oxide by reducing its oxidative inactivation. J. Hypertens. 2005, 23, 589–596.
160. Sorrentino, S.A.; Doerries, C.; Manes, C.; Speer, T.; Dessy, C.; Lobysheva, I.; Mohmand, W.;
Akbar, R.; Bahlmann, F.; Besler, C.; et al. Nebivolol exerts beneficial effects on endothelial
function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular
dysfunction early after myocardial infarction beyond conventional β1-blockade. J. Am. Coll. Cardiol.
2011, 57, 601–611.
161. Munzel, T.; Daiber, A.; Mulsch, A. Explaining the phenomenon of nitrate tolerance. Circ. Res.
2005, 97, 618–628.
162. Gori, T.; Daiber, A. Non-hemodynamic effects of organic nitrates and the distinctive
characteristics of pentaerithrityl tetranitrate. Am. J. Cardiovasc. Drugs. 2009, 9, 7–15.
Int. J. Mol. Sci. 2015, 16
163. Munzel, T.; Meinertz, T.; Tebbe, U.; Schneider, H.T.; Stalleicken, D.; Wargenau, M.; Gori, T.;
Klingmann, I.; Investigators, C.S. Efficacy of the long-acting nitro vasodilator pentaerithrityl
tetranitrate in patients with chronic stable angina pectoris receiving anti-anginal background
therapy with β-blockers: A 12-week, randomized, double-blind, placebo-controlled trial.
Eur. Heart. J. 2014, 35, 895–903.
164. Schnorbus, B.; Schiewe, R.; Ostad, M.A.; Medler, C.; Wachtlin, D.; Wenzel, P.; Daiber, A.;
Munzel, T.; Warnholtz, A. Effects of pentaerythritol tetranitrate on endothelial function in
coronary artery disease: Results of the penta study. Clin. Res. Cardiol. 2010, 99, 115–124.
165. Schuhmacher, S.; Wenzel, P.; Schulz, E.; Oelze, M.; Mang, C.; Kamuf, J.; Gori, T.; Jansen, T.;
Knorr, M.; Karbach, S.; et al. Pentaerythritol tetranitrate improves angiotensin II-induced
vascular dysfunction via induction of heme oxygenase-1. Hypertension 2010, 55, 897–904.
166. Wenzel, P.; Oelze, M.; Coldewey, M.; Hortmann, M.; Seeling, A.; Hink, U.; Mollnau, H.;
Stalleicken, D.; Weiner, H.; Lehmann, J.; et al. Heme oxygenase-1: A novel key player in the
development of tolerance in response to organic nitrates. Arterioscler. Thromb. Vasc. Biol. 2007,
27, 1729–1735.
167. Oberle, S.; Abate, A.; Grosser, N.; Hemmerle, A.; Vreman, H.J.; Dennery, P.A.; Schneider, H.T.;
Stalleicken, D.; Schroder, H. Endothelial protection by pentaerithrityl trinitrate: Bilirubin and
carbon monoxide as possible mediators. Exp. Biol. Med. (Maywood) 2003, 228, 529–534.
168. Oberle, S.; Abate, A.; Grosser, N.; Vreman, H.J.; Dennery, P.A.; Schneider, H.T.; Stalleicken, D.;
Schroder, H. Heme oxygenase-1 induction may explain the antioxidant profile of pentaerythrityl
trinitrate. Biochem. Biophys. Res. Commun. 2002, 290, 1539–1544.
169. Schuhmacher, S.; Oelze, M.; Bollmann, F.; Kleinert, H.; Otto, C.; Heeren, T.; Steven, S.;
Hausding, M.; Knorr, M.; Pautz, A.; et al. Vascular dysfunction in experimental diabetes is
improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 2011,
60, 2608–2616.
170. Oppermann, M.; Balz, V.; Adams, V.; Dao, V.T.; Bas, M.; Suvorava, T.; Kojda, G.
Pharmacological induction of vascular extracellular superoxide dismutase expression in vivo.
J. Cell. Mol. Med. 2009, 13, 1271–1278.
171. Hacker, A.; Muller, S.; Meyer, W.; Kojda, G. The nitric oxide donor pentaerythritol tetranitrate
can preserve endothelial function in established atherosclerosis. Br. J. Pharmacol. 2001, 132,
1707–1714.
172. Flierl, U.; Fraccarollo, D.; Widder, J.D.; Micka, J.; Neuser, J.; Bauersachs, J.; Schafer, A. The
nitric oxide donor pentaerythritol tetranitrate reduces platelet activation in congestive heart
failure. PLoS ONE 2015, 10, e0123621.
173. Thum, T.; Fraccarollo, D.; Thum, S.; Schultheiss, M.; Daiber, A.; Wenzel, P.; Munzel, T.;
Ertl, G.; Bauersachs, J. Differential effects of organic nitrates on endothelial progenitor cells are
determined by oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 748–754.
174. Thum, T.; Wiebking, V.; Ertl, G.; Bauersachs, J. Organic nitrates differentially modulate
circulating endothelial progenitor cells and endothelial function in patients with symptomatic
coronary artery disease. Antioxid. Redox Signal. 2011, 15, 925–931.
Int. J. Mol. Sci. 2015, 16
175. DiFabio, J.M.; Thomas, G.R.; Zucco, L.; Kuliszewski, M.A.; Bennett, B.M.; Kutryk, M.J.;
Parker, J.D. Nitroglycerin attenuates human endothelial progenitor cell differentiation, function,
and survival. J. Pharmacol. Exp. Ther. 2006, 318, 117–123.
176. Pautz, A.; Rauschkolb, P.; Schmidt, N.; Art, J.; Oelze, M.; Wenzel, P.; Forstermann, U.; Daiber, A.;
Kleinert, H. Effects of nitroglycerin or pentaerithrityl tetranitrate treatment on the gene expression
in rat hearts: Evidence for cardiotoxic and cardioprotective effects. Physiol. Genom. 2009, 38,
176–185.
177. Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H.
Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces
blood pressure in female offspring. Hypertension 2015, 65, 232–237.
178. Abbott, C.A.; Yu, D.M.; Woollatt, E.; Sutherland, G.R.; McCaughan, G.W.; Gorrell, M.D.
Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP)
IV homolog, DPP8. Eur. J. Biochem. 2000, 267, 6140–6150.
179. Zhong, J.; Rao, X.; Rajagopalan, S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond
glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013, 226,
305–314.
180. Buhling, F.; Junker, U.; Reinhold, D.; Neubert, K.; Jager, L.; Ansorge, S. Functional role of
CD26 on human B lymphocytes. Immunol. Lett. 1995, 45, 47–51.
181. Buhling, F.; Kunz, D.; Reinhold, D.; Ulmer, A.J.; Ernst, M.; Flad, H.D.; Ansorge, S. Expression
and functional role of dipeptidyl peptidase IV (CD26) on human natural killer cells. Nat. Immun.
1994, 13, 270–279.
182. Gorrell, M.D. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders.
Clin. Sci. 2005, 108, 277–292.
183. Palmieri, F.E.; Ward, P.E. Dipeptidyl(amino)peptidase iv and post proline cleaving enzyme in
cultured endothelial and smooth muscle cells. Adv. Exp. Med. Biol. 1989, 247A, 305–311.
184. Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132,
185. Lund, P.K.; Goodman, R.H.; Dee, P.C.; Habener, J.F. Pancreatic preproglucagon cdna contains
two glucagon-related coding sequences arranged in tandem. Proc. Natl. Acad. Sci. USA 1982, 79,
345–349.
186. Doyle, M.E.; Egan, J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas.
Pharmacol. Ther. 2007, 113, 546–593.
187. Nauck, M.A. Incretin-based therapies for type 2 diabetes mellitus: Properties, functions, and
clinical implications. Am. J. Med. 2011, 124, S3–18.
188. Brubaker, P.L.; Drucker, D.J. Structure-function of the glucagon receptor family of G
protein-coupled receptors: The glucagon, GIP, GLP-1, and GLP-2 receptors. Recept. Channels
2002, 8, 179–188.
189. Mentlein, R.; Gallwitz, B.; Schmidt, W.E. Dipeptidyl-peptidase iv hydrolyses gastric inhibitory
polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is
responsible for their degradation in human serum. Eur. J. Biochem. 1993, 214, 829–835.
Int. J. Mol. Sci. 2015, 16
190. Kieffer, T.J.; McIntosh, C.H.; Pederson, R.A. Degradation of glucose-dependent insulinotropic
polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV.
Endocrinology 1995, 136, 3585–3596.
191. International Diabetes Federation Guideline Development Group. Global guideline for type 2
diabetes. Diabetes Res. Clin. Pract. 2014, 104, 1–52.
192. Matsubara, J.; Sugiyama, S.; Sugamura, K.; Nakamura, T.; Fujiwara, Y.; Akiyama, E.;
Kurokawa, H.; Nozaki, T.; Ohba, K.; Konishi, M.; et al. A dipeptidyl peptidase-4 inhibitor,
des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation
in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 2012, 59, 265–276.
193. Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.;
Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces
atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis.
Circulation 2011, 124, 2338–2349.
194. Nishioka, T.; Shinohara, M.; Tanimoto, N.; Kumagai, C.; Hashimoto, K. Sitagliptin, a dipeptidyl
peptidase-iv inhibitor, improves psoriasis. Dermatology 2012, 224, 20–21.
195. Kern, M.; Kloting, N.; Niessen, H.G.; Thomas, L.; Stiller, D.; Mark, M.; Klein, T.; Bluher, M.
Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PLoS ONE
2012, 7, e38744.
196. Darsalia, V.; Ortsater, H.; Olverling, A.; Darlof, E.; Wolbert, P.; Nystrom, T.; Klein, T.;
Sjoholm, A.; Patrone, C. The DPP-4 inhibitor linagliptin counteracts stroke in the normal and
diabetic mouse brain: A comparison with glimepiride. Diabetes 2013, 62, 1289–1296.
197. Huang, C.Y.; Shih, C.M.; Tsao, N.W.; Lin, Y.W.; Huang, P.H.; Wu, S.C.; Lee, A.W.; Kao, Y.T.;
Chang, N.C.; Nakagami, H.; et al. Dipeptidyl peptidase-4 inhibitor improves neovascularization
by increasing circulating endothelial progenitor cells. Br. J. Pharmacol. 2012, 167, 1506–1519.
198. Brenner, C.; Krankel, N.; Kuhlenthal, S.; Israel, L.; Remm, F.; Fischer, C.; Herbach, N.;
Speer, T.; Grabmaier, U.; Laskowski, A.; et al. Short-term inhibition of DPP-4 enhances
endothelial regeneration after acute arterial injury via enhanced recruitment of circulating
progenitor cells. Int. J. Cardiol. 2014, 177, 266–275.
199. Fadini, G.P.; Boscaro, E.; Albiero, M.; Menegazzo, L.; Frison, V.; de Kreutzenberg, S.;
Agostini, C.; Tiengo, A.; Avogaro, A. The oral dipeptidyl peptidase-4 inhibitor sitagliptin
increases circulating endothelial progenitor cells in patients with type 2 diabetes: Possible role of
stromal-derived factor-1α. Diabetes care 2010, 33, 1607–1609.
200. Xiao-Yun, X.; Zhao-Hui, M.; Ke, C.; Hong-Hui, H.; Yan-Hong, X. Glucagon-like peptide-1
improves proliferation and differentiation of endothelial progenitor cells via upregulating vegf
generation. Med. Sci. Monit. 2011, 17, BR35–BR41.
201. Oeseburg, H.; de Boer, R.A.; Buikema, H.; van der Harst, P.; van Gilst, W.H.; Sillje, H.H.
Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence
through the activation of protein kinase a. Arterioscler. Thromb. Vasc. biol. 2010, 30, 1407–1414.
202. Akarte, A.S.; Srinivasan, B.P.; Gandhi, S.; Sole, S. Chronic DPP-IV inhibition with PKF-275-055
attenuates inflammation and improves gene expressions responsible for insulin secretion in
streptozotocin induced diabetic rats. Eur. J. Pharm. Sci. 2012, 47, 456–463.
Int. J. Mol. Sci. 2015, 16
203. Chinda, K.; Palee, S.; Surinkaew, S.; Phornphutkul, M.; Chattipakorn, S.; Chattipakorn, N.
Cardioprotective effect of dipeptidyl peptidase-4 inhibitor during ischemia-reperfusion injury.
Int. J. Cardiol. 2013, 167, 451–457.
204. Inthachai, T.; Lekawanvijit, S.; Kumfu, S.; Apaijai, N.; Pongkan, W.; Chattipakorn, S.C.;
Chattipakorn, N. Dipeptidyl peptidase-4 inhibitor improves cardiac function by attenuating
adverse cardiac remodelling in rats with chronic myocardial infarction. Exp. Physiol. 2015, 100,
667–679.
205. Bao, W.; Morimoto, K.; Hasegawa, T.; Sasaki, N.; Yamashita, T.; Hirata, K.; Okita, Y.; Okada, K.
Orally administered dipeptidyl peptidase-4 inhibitor (alogliptin) prevents abdominal aortic
aneurysm formation through an antioxidant effect in rats. J. Vasc. Surg. 2014, 59, 1098–1108.
206. Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone
parkinson's disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways.
J. Neurochem. 2015, 133, 700–707.
207. Steven, S.; Hausding, M.; Kroller-Schon, S.; Mader, M.; Mikhed, Y.; Stamm, P.; Zinssius, E.;
Pfeffer, A.; Welschof, P.; Agdauletova, S.; et al. Gliptin and GLP-1 analog treatment improves
survival and vascular inflammation/dysfunction in animals with lipopolysaccharide-induced
endotoxemia. Basic Res. Cardiol. 2015, 110, 6.
208. Kroller-Schon, S.; Knorr, M.; Hausding, M.; Oelze, M.; Schuff, A.; Schell, R.; Sudowe, S.;
Scholz, A.; Daub, S.; Karbach, S.; et al. Glucose-independent improvement of vascular dysfunction
in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc. Res. 2012, 96, 140–149.
209. Shah, P.; Ardestani, A.; Dharmadhikari, G.; Laue, S.; Schumann, D.M.; Kerr-Conte, J.; Pattou, F.;
Klein, T.; Maedler, K. The DPP-4 inhibitor linagliptin restores β-cell function and survival
in human isolated islets through GLP-1 stabilization. J. Clin. Endocrinol. Metab. 2013, 98,
E1163–E1172.
210. Rizzo, M.R.; Barbieri, M.; Marfella, R.; Paolisso, G. Reduction of oxidative stress and
inflammation by blunting daily acute glucose fluctuations in patients with type 2 diabetes: Role
of dipeptidyl peptidase-iv inhibition. Diabetes Care 2012, 35, 2076–2082.
211. Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.;
Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via
inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis
2014, 232, 156–164.
212. Shiraki, A.; Oyama, J.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.;
Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide
reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis
2012, 221, 375–382.
213. Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of
glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular
disorders. Br. J. Pharmacol. 2012, 166, 1586–1599.
214. Fisslthaler, B.; Fleming, I. Activation and signaling by the amp-activated protein kinase in
endothelial cells. Circ. Res. 2009, 105, 114–127.
Int. J. Mol. Sci. 2015, 16
215. Balteau, M.; van Steenbergen, A.; Timmermans, A.D.; Dessy, C.; Behets-Wydemans, G.;
Tajeddine, N.; Castanares-Zapatero, D.; Gilon, P.; Vanoverschelde, J.L.; Horman, S.; et al.
AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by
hyperglycemia in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 307,
H1120–H1133.
216. Martin, M.; Huguet, J.; Centelles, J.J.; Franco, R. Expression of ecto-adenosine deaminase and
CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine
deaminase as costimulatory molecule. J. Immunol. 1995, 155, 4630–4643.
217. Kameoka, J.; Tanaka, T.; Nojima, Y.; Schlossman, S.F.; Morimoto, C. Direct association of
adenosine deaminase with a T cell activation antigen, CD26. Science 1993, 261, 466–469.
218. Dopheide, J.F.; Scheer, M.; Doppler, C.; Obst, V.; Stein, P.; Vosseler, M.; Abegunewardene, N.;
Gori, T.; Munzel, T.; Daiber, A.; et al. Change of walking distance in intermittent claudication:
Impact on inflammation, oxidative stress and mononuclear cells: A pilot study. Clin. Res. Cardiol.
2015, doi:10.1007/s00392-015-0840-5.
219. Dopheide, J.F.; Obst, V.; Doppler, C.; Radmacher, M.C.; Scheer, M.; Radsak, M.P.; Gori, T.;
Warnholtz, A.; Fottner, C.; Daiber, A.; et al. Phenotypic characterisation of pro-inflammatory
monocytes and dendritic cells in peripheral arterial disease. Thromb. Haemost. 2012, 108,
1198–1207.
220. Dopheide, J.F.; Doppler, C.; Scheer, M.; Obst, V.; Radmacher, M.C.; Radsak, M.P.; Gori, T.;
Warnholtz, A.; Fottner, C.; Munzel, T.; et al. Critical limb ischaemia is characterised by an
increased production of whole blood reactive oxygen species and expression of TREM-1 on
neutrophils. Atherosclerosis 2013, 229, 396–403.
221. Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051.
222. Burgmaier, M.; Liberman, A.; Mollmann, J.; Kahles, F.; Reith, S.; Lebherz, C.; Marx, N.;
Lehrke, M. Glucagon-like peptide-1 (GLP-1) and its split products GLP-1(9-37) and GLP-1(28-37)
stabilize atherosclerotic lesions in apoe−/− mice. Atherosclerosis 2013, 231, 427–435.
223. Gaspari, T.; Liu, H.; Welungoda, I.; Hu, Y.; Widdop, R.E.; Knudsen, L.B.; Simpson, R.W.;
Dear, A.E. A GLP-1 receptor agonist liraglutide inhibits endothelial cell dysfunction and
vascular adhesion molecule expression in an apoe−/− mouse model. Diabetes Vasc. Dis. Res.
2011, 8, 117–124.
224. Buldak, L.; Labuzek, K.; Buldak, R.J.; Machnik, G.; Boldys, A.; Okopien, B. Exenatide (a GLP-1
agonist) improves the antioxidative potential of in vitro cultured human monocytes/macrophages.
Naunyn Schmiedebergs Arch. Pharmacol. 2015, in press.
225. Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.;
Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation
of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010,
59, 1030–1037.
226. Erdogdu, O.; Nathanson, D.; Sjoholm, A.; Nystrom, T.; Zhang, Q. Exendin-4 stimulates
proliferation of human coronary artery endothelial cells through eNOS-, PKA- and
PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol. Cell. Endocrinol. 2010, 325,
26–35.
Int. J. Mol. Sci. 2015, 16
227. Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.S.; Drucker, D.J.; Husain, M. Cardioprotective
and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both
glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008, 117,
2340–2350.
228. Green, B.D.; Hand, K.V.; Dougan, J.E.; McDonnell, B.M.; Cassidy, R.S.; Grieve, D.J. GLP-1
and related peptides cause concentration-dependent relaxation of rat aorta through a pathway
involving katp and camp. Arch. Biochem. Biophys. 2008, 478, 136–142.
229. Shah, Z.; Pineda, C.; Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Racoma, I.; Deiuliis, J.; Xu, X.;
Sun, Q.; Moffatt-Bruce, S.; et al. Acute DPP-4 inhibition modulates vascular tone through GLP-1
independent pathways. Vascul. Pharmacol. 2011, 55, 2–9.
230. Wang, R.; Lu, L.; Guo, Y.; Lin, F.; Chen, H.; Chen, W.; Chen, M. Effect of glucagon-like
peptide-1 on high-glucose-induced oxidative stress and cell apoptosis in human endothelial
cells and its underlying mechanism. J. Cardiovasc. Pharmacol. 2015, doi:10.1097/
FJC.0000000000000255.
231. Levy, M.M.; Artigas, A.; Phillips, G.S.; Rhodes, A.; Beale, R.; Osborn, T.; Vincent, J.L.;
Townsend, S.; Lemeshow, S.; Dellinger, R.P. Outcomes of the surviving sepsis campaign in
intensive care units in the USA and europe: A prospective cohort study. Lancet Infect. Dis. 2012,
12, 919–924.
232. Levi, M.; van der Poll, T. Disseminated intravascular coagulation: A review for the internist.
Intern. Emerg. Med. 2013, 8, 23–32.
233. Boerma, E.C.; van der Voort, P.H.; Spronk, P.E.; Ince, C. Relationship between sublingual and
intestinal microcirculatory perfusion in patients with abdominal sepsis. Crit. Care Med. 2007, 35,
1055–1060.
234. Bone, R.C. Sepsis syndrome. New insights into its pathogenesis and treatment. Infect. Dis. Clin.
N. Am. 1991, 5, 793–805.
235. Nguyen, H.B.; Rivers, E.P.; Knoblich, B.P.; Jacobsen, G.; Muzzin, A.; Ressler, J.A.;
Tomlanovich, M.C. Early lactate clearance is associated with improved outcome in severe sepsis
and septic shock. Crit. Care Med. 2004, 32, 1637–1642.
236. Gkaliagkousi, E.; Ferro, A. Nitric oxide signalling in the regulation of cardiovascular and platelet
function. Front. Biosci. 2011, 16, 1873–1897.
237. Huet, O.; Dupic, L.; Harrois, A.; Duranteau, J. Oxidative stress and endothelial dysfunction
during sepsis. Front. Biosci. 2011, 16, 1986–1995.
238. Krotz, F.; Sohn, H.Y.; Pohl, U. Reactive oxygen species: Players in the platelet game.
Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1988–1996.
239. Heyworth, P.G.; Cross, A.R.; Curnutte, J.T. Chronic granulomatous disease. Curr. Opin. Immunol.
2003, 15, 578–584.
240. Nathens, A.B.; Neff, M.J.; Jurkovich, G.J.; Klotz, P.; Farver, K.; Ruzinski, J.T.; Radella, F.;
Garcia, I.; Maier, R.V. Randomized, prospective trial of antioxidant supplementation in critically
ill surgical patients. Ann. Surg. 2002, 236, 814–822.
241. Domenighetti, G.; Suter, P.M.; Schaller, M.D.; Ritz, R.; Perret, C. Treatment with N-acetylcysteine
during acute respiratory distress syndrome: A randomized, double-blind, placebo-controlled
clinical study. J. Crit. Care 1997, 12, 177–182.
Int. J. Mol. Sci. 2015, 16
242. Sprung, C.L.; Annane, D.; Keh, D.; Moreno, R.; Singer, M.; Freivogel, K.; Weiss, Y.G.;
Benbenishty, J.; Kalenka, A.; Forst, H.; et al. Hydrocortisone therapy for patients with septic
shock. N. Engl. J. Med. 2008, 358, 111–124.
243. National Heart, L.; Blood Institute, A.C.T.N.; Truwit, J.D.; Bernard, G.R.; Steingrub, J.;
Matthay, M.A.; Liu, K.D.; Albertson, T.E.; Brower, R.G.; Shanholtz, C.; et al. Rosuvastatin for
sepsis-associated acute respiratory distress syndrome. N. Engl. J. Med. 2014, 370, 2191–2200.
244. Ku, H.C.; Chen, W.P.; Su, M.J. GLP-1 signaling preserves cardiac function in endotoxemic
fischer 344 and DPP4-deficient rats. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 382,
463–474.
245. Amori, R.E.; Lau, J.; Pittas, A.G. Efficacy and safety of incretin therapy in type 2 diabetes:
Systematic review and meta-analysis. JAMA 2007, 298, 194–206.
246. Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; van der Tuin, S.J.; Zhang, H.; Bieghs, V.; Jawad, A.H.;
Shiri-Sverdlov, R.; Bot, I.; de Jager, S.C.; et al. Exendin-4 decreases liver inflammation and
atherosclerosis development simultaneously by reducing macrophage infiltration. Br. J. Pharmacol.
2014, 171, 723–734.
247. Jung, Y.A.; Choi, Y.K.; Jung, G.S.; Seo, H.Y.; Kim, H.S.; Jang, B.K.; Kim, J.G.; Lee, I.K.;
Kim, M.K.; Park, K.G. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis.
Diabetes Res. Clin. Pract. 2014, 105, 47–57.
248. Weng, S.Y.; Schuppan, D. AMPK regulates macrophage polarization in adipose tissue
inflammation and NASH. J. Hepatol. 2013, 58, 619–621.
249. Iwaya, H.; Fujii, N.; Hagio, M.; Hara, H.; Ishizuka, S. Contribution of dipeptidyl peptidase IV to the
severity of dextran sulfate sodium-induced colitis in the early phase. Biosci. Biotechnol. Biochem.
2013, 77, 1461–1466.
250. Schade, J.; Schmiedl, A.; Kehlen, A.; Veres, T.Z.; Stephan, M.; Pabst, R.; von Horsten, S.
Airway-specific recruitment of T cells is reduced in a CD26-deficient F344 rat substrain.
Clin. Exp. Immunol. 2009, 158, 133–142.
251. Viby, N.E.; Isidor, M.S.; Buggeskov, K.B.; Poulsen, S.S.; Hansen, J.B.; Kissow, H.
Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of
experimental obstructive lung disease in female mice. Endocrinology 2013, 154, 4503–4511.
252. Busso, N.; Wagtmann, N.; Herling, C.; Chobaz-Peclat, V.; Bischof-Delaloye, A.; So, A.;
Grouzmann, E. Circulating CD26 is negatively associated with inflammation in human and
experimental arthritis. Am. J. Pathol. 2005, 166, 433–442.
253. Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.;
Braunwald, E.; Pravastatin or Atorvastatin, E.; Infection Therapy-Thrombolysis in Myocardial
Infarction, I. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005,
352, 20–28.
254. Zhu, F.; Wang, Q.; Guo, C.; Wang, X.; Cao, X.; Shi, Y.; Gao, F.; Ma, C.; Zhang, L. IL-17
induces apoptosis of vascular endothelial cells: A potential mechanism for human acute coronary
syndrome. Clin. Immunol. 2011, 141, 152–160.
255. Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.;
Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced
blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034.
Int. J. Mol. Sci. 2015, 16
256. Roussel, L.; Houle, F.; Chan, C.; Yao, Y.; Berube, J.; Olivenstein, R.; Martin, J.G.; Huot, J.;
Hamid, Q.; Ferri, L.; et al. IL-17 promotes p38 MAPK-dependent endothelial activation
enhancing neutrophil recruitment to sites of inflammation. J. Immunol. 2010, 184, 4531–4537.
257. Hoch, N.E.; Guzik, T.J.; Chen, W.; Deans, T.; Maalouf, S.A.; Gratze, P.; Weyand, C.;
Harrison, D.G. Regulation of t-cell function by endogenously produced angiotensin II. Am. J.
Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R208–R216.
258. Geiger, H. T-cells in angiotensin-II-induced vascular damage. Nephrol. Dial. Transplant. 2008,
23, 1107–1108.
259. Micha, R.; Imamura, F.; Wyler von Ballmoos, M.; Solomon, D.H.; Hernan, M.A.; Ridker, P.M.;
Mozaffarian, D. Systematic review and meta-analysis of methotrexate use and risk of
cardiovascular disease. Am. J. Cardiol. 2011, 108, 1362–1370.
260. Asanuma, H.; Sanada, S.; Ogai, A.; Minamino, T.; Takashima, S.; Asakura, M.; Ogita, H.;
Shinozaki, Y.; Mori, H.; Node, K.; et al. Methotrexate and MX-68, a new derivative of
methotrexate, limit infarct size via adenosine-dependent mechanisms in canine hearts.
J. Cardiovasc. Pharmacol. 2004, 43, 574–579.
261. Moreira, D.M.; Lueneberg, M.E.; da Silva, R.L.; Fattah, T.; Mascia Gottschall, C.A. Rationale
and design of the tethys trial: The effects of methotrexate therapy on myocardial infarction with
ST-segment elevation. Cardiology 2013, 126, 167–170.
262. Everett, B.M.; Pradhan, A.D.; Solomon, D.H.; Paynter, N.; Macfadyen, J.; Zaharris, E.;
Gupta, M.; Clearfield, M.; Libby, P.; Hasan, A.A.; et al. Rationale and design of the
cardiovascular inflammation reduction trial: A test of the inflammatory hypothesis of
atherothrombosis. Am. Heart J. 2013, 166, 199–207 e115.
263. Elhage, R.; Maret, A.; Pieraggi, M.T.; Thiers, J.C.; Arnal, J.F.; Bayard, F. Differential effects of
interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak
formation in apolipoprotein E-deficient mice. Circulation 1998, 97, 242–244.
264. Chi, H.; Messas, E.; Levine, R.A.; Graves, D.T.; Amar, S. Interleukin-1 receptor signaling
mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine
apolipoprotein e heterozygote model: Pharmacotherapeutic implications. Circulation 2004, 110,
1678–1685.
265. Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T.;
Group, C.P.I. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids,
C-reactive protein, interleukin-6, and fibrinogen: A phase IIB randomized, placebo-controlled
trial. Circulation 2012, 126, 2739–2748.
266. Munzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and
prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196.
267. Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in
cardiovascular disease? Eur. Heart. J. 2010, 31, 2741–2748.
268. Zhang, L.; Zalewski, A.; Liu, Y.; Mazurek, T.; Cowan, S.; Martin, J.L.; Hofmann, S.M.;
Vlassara, H.; Shi, Y. Diabetes-induced oxidative stress and low-grade inflammation in porcine
coronary arteries. Circulation 2003, 108, 472–478.
269. Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101,
Int. J. Mol. Sci. 2015, 16
270. Warnholtz, A.; Genth-Zotz, S.; Munzel, T. Should treatment of sepsis include statins?
Circulation 2005, 111, 1735–1737.
271. Merx, M.W.; Liehn, E.A.; Janssens, U.; Lutticken, R.; Schrader, J.; Hanrath, P.; Weber, C.
HMG-CoA reductase inhibitor simvastatin profoundly improves survival in a murine model of
sepsis. Circulation 2004, 109, 2560–2565.
272. Deshpande, A.; Pasupuleti, V.; Rothberg, M.B. Statin therapy and mortality from sepsis: A
meta-analysis of randomized trials. Am. J. Med. 2015, 128, 410–417 e411.
2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Source: http://eu-ros.eu/wp-content/uploads/2016/04/2015-Steven-Review-Antioxidants2_0-DPP4-GLP1-IJMS.pdf
r ers une politique médicaments au Canada Marc-André Gagnon, Ph. D. Publié par :La Fédération canadienne des syndicats d'infirmières et infirmierswww.fcsii.ca2841, promenade RiversideOttawa (Ontario) K1V 8X7613-526-4661 © La Fédération canadienne des syndicats d'infirmières et infirmiers 2014 Tous droits réservés. Aucune partie de cet ouvrage ne peut pas être reproduite ou transmise par quelque procédé que ce soit, tant électronique que mécanique, en particulier par photocopie, enregistrement ou par tout système de recherché ou d'entreposage documentaire sans l'autorisation de l'éditeur.
UTAH 4-H PROGRAM GUIDE UTAH 4-H LEADERSHIP Developing leadership skills is one of the most important goals of 4-H and key to positive youth development. Opportunities for leadership development are naturally found throughout all 4-H programs. As youth engage in 4-H programs they learn skills such as public speaking, planning and decision making, facilitation techniques, and teamwork. When youth become competent leaders, they gain the confidence they need to make decisions that affect them now and throughout their lives.



