Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Dottorato.dsf.unica.it
J Mol ModelDOI 10.1007/s00894-010-0698-4
Investigating reaction pathways in rare events simulationsof antibiotics diffusion through protein channels
Eric Hajjar & Amit Kumar & Paolo Ruggerone &Matteo Ceccarelli
Received: 1 December 2009 / Accepted: 27 February 2010
# Springer-Verlag 2010
Abstract In Gram-negative bacteria, outer-membrane pro-
further simulations. This will benefit the screening and design
tein channels, such as OmpF of Escherichia coli, constitute
for antibiotics with better permeation properties.
the entry point of various classes of antibiotics. Whileantibacterial research and development is declining, bacterial
Keywords Accelerated molecular dynamics . Antibacterial
resistance to antibiotics is rising and there is an emergency
design . Antibiotics uptake . Bacterial porins . Diffusion .
call for a new way to develop potent antibacterial agents and
Free energy . Reaction coordinate
to bring them to the market faster and at reduced cost. Anemerging strategy is to follow a bottom-up approach based onmicroscopically founded computational based screening,
however such strategy needs better-tuned methods. Here wepropose to use molecular dynamics (MD) simulations
The permeability to antibiotics, or uptake, is the very first
combined with the metadynamics algorithm, to study
line of defense of Gram-negative bacteria, that are
antibiotic translocation through OmpF at a molecular scale.
protected by an outer-membrane []. In the case of E.coli,
This recently designed algorithm overcomes the time scale
the uptake of several classes of β-lactam antibiotics, a
problem of classical MD by accelerating some reaction
prominent group in our current antibacterial arsenal, is
coordinates. It is expected that the initial assumption of the
largely controlled by general diffusion protein channels
reaction coordinates is a key determinant for the efficiency
such as outer membrane protein F (OmpF) [Indeed,
and accuracy of the simulations. Previous studies using
pathogenic strains of Gram-negative bacteria, that were
different computational schemes for a similar process only
found to be resistant against quinolones and β-lactams
used one reaction coordinate, which is the directionality. Here
(two of the main classes of antibiotics) frequently have
we go further and see how it is possible to include more
modulation of the structure or the expression of general
informative reaction coordinates, accounting explicitly for:
diffusion porin OmpF
(i) the antibiotic flexibility and (ii) interactions with the
A key feature in the structure of porins, as seen from the
channel. As model systems, we select two compounds
X-ray structure of OmpF is the presence of the loop L3
covering the main classes of antibiotics, ampicillin and
that folds back into the channel to form a gate, also called
moxifloxacine. We decipher the molecular mechanism of
constriction region (see Fig. In addition to such spatial
translocation of each antibiotic and highlight the important
constriction, the zone is also characterized by a strong
parameters that should be taken into account for improving
transversal electric field, generated by negatively chargedresidues D113, E117 (L3 side) that faces a cluster ofpositively charged residues R42, R82, and R132 (anti-L3
E. Hajjar (
*) : A. Kumar : P. Ruggerone : M. Ceccarelli
side) (see Fig. ).
Department of Physics and UOS-SLACS,
While antibacterial research and development is on the
Universita di Cagliari and Istituto Officina dei Materiali/CNR,
decline, the resistance is on the rise, and we are thus facing
SP Monserrato-Sestu Km 0.700,
an alarming situation where there is an emergency call for a
I-09042 Monserrato, Italye-mail:
[email protected]
new way to develop potent antibacterial agents and to bring
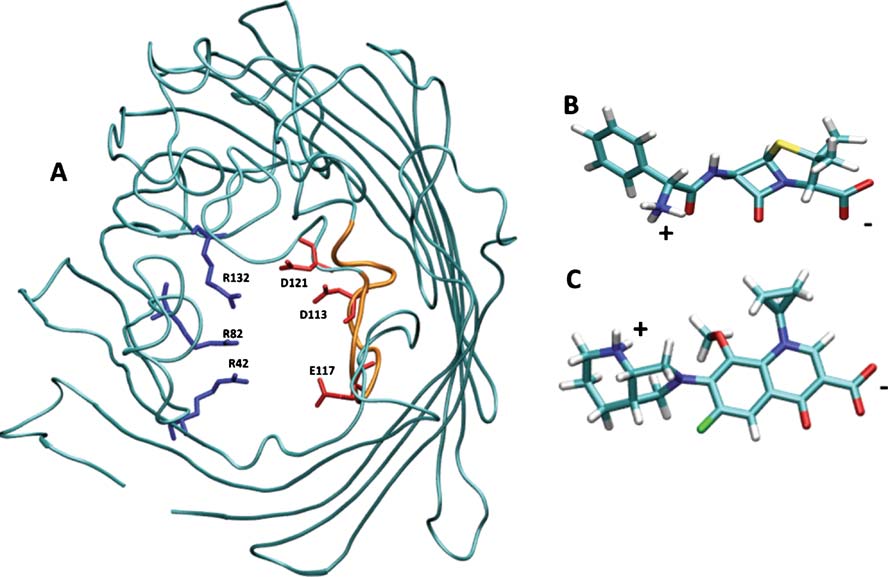
Fig. 1 a) Structural details of OmpF. The backbone of OmpF is
type (positively charged in blue, negatively charged in red). The loop
displayed in cyan cartoons to highlight its secondary structure. The
L3 is colored in orange. 3D structures of the optimized geometry of
charged residues at the constriction region (D113, E117, D121 on the
ampicillin (b) and (c) moxifloxacin. The molecules are colored by atom
L3 side and R42, R82, R132 on the anti-L3 side) are colored by residue
types: blue for nitrogen, red for oxygen, cyan for carbon
them to the market faster and at reduced cost. An emerging
specifying a single reaction coordinate can lead to a strong
strategy is to follow a bottom-up approach, from the
approximation of the sampled process.
knowledge of resistant mechanisms to a rational structure-
Several studies addressed the challenge of such biased
based design and screening of antibiotics. Within this
molecular dynamics methods to study the difficult problem
scheme, molecular simulations have the potential to provide
of molecular diffusion through narrow channels. In their
an accurate microscopic explanation of what governs
recent paper, Henin et al. highlight the importance of
antibiotics diffusion (and thus bacterial resistance) and
using another reaction coordinates than the usually taken, Z
how to screen for better antibiotics. In principle, standard
(distance) coordinate that drives the process under study.
MD simulations would have the required microscopic
Indeed it is believed that including an extra reaction
accuracy to link the structure and dynamics (of the drug
coordinate would allow explicitly to account for reorienta-
and porin) to the rate of permeation. However, standard
tions and/or relaxation needed for reaching better accuracy
simulations are limited to hundred of nanoseconds at most
in the sampling of the process studied. In fact, our and
and they do not allow the study of the reactive pathway that
others previous studies suggested that the flexibility and
antibiotics follow during passive diffusion, which is on the
orientation of the antibiotic would play a role in its
order of hundreds of microseconds
diffusion process through OmpF [, However, the
To overcome this timescale problem, we propose to use
qualitative and quantitative consequences of the choice of
accelerated MD simulation algorithms schemes, or metady-
RC are still poorly understood and there are very few
namics, while keeping an "all atom" description of the
studies illustrating this point. The first natural reaction
systems The metadynamics algorithm (to accelerate
coordinate that could be used in our metadynamics
sampling) is based on the following principle: a time-
approach is the position of the antibiotic with respect to
dependent bias is added on a few chosen reaction coor-
the axis of diffusion Z.
dinates (RC). It is important to note that a crucial point of
Here we go further and include more informative
such metadynamic approach is the choice of appropriate RC.
reaction coordinates, accounting not only for the direction-
Unlike other popular techniques (such as steered
ality of the transport but also explicitly for: (i) the antibiotic
molecular dynamics metadynamics allows more than
orientation and (ii) specific interactions with the channel.
one RC to be defined and this is an important advantage as
Our approach will allow discussing the two choices of RC
in the translocation with the study of two different
for system setup and simulation [All simulated systems
antibiotics: the beta-lactam ampicillin and the quinolone
were validated for convergence and stabilization of energy,
moxifloxacine, for which experimental and simulation data
temperature and root mean square deviation with respect to
are already available [].
the starting structure.
Finally, we conclude by discussing the improved
strategies, as highlighted by our study. Indeed, the in-
Metadynamics algorithm
depth analysis of these two model systems allows us todiscuss for future improvements in the biased methods
The metadynamics algorithm employs a bias to accelerate
(importance of explicitly including the relative orientation
the evolution of some collective variables, defined as the
and the specific interactions) to screen and design for
relevant reaction coordinates for the process under investi-
antibiotics in particular, but also for investigating any other
gation. The bias consists of a history dependent potential,
complex process of interest.
which is constructed as a sum of repulsive potentialcentered along the trajectory of the collective variables.
These additional energy terms avoid revisiting the same
Materials and methods
conformations or at least add a penalty term to thepreviously visited conformations. In the present simula-
Starting structures for molecular dynamic simulations
tions, a Gaussian potential is added every 4 ps with a heightof 1.0 kJ mol−1. The Gaussian width is set to 0.2 Å, 5.0
We followed the same protocol of simulations as described
degrees and 0.5, respectively for the distance Z, the angle Θ
earlier starting from the crystal structure ‘2OMF' (pdb-
and the number of hydrogen bonds. These parameters were
code) and residues protonation state as in [We added
chosen to allow a better resolution in the sampling of the
the required amount of Cl- and K+ counter ions to neutralize
free energy and a low error (around 2kBT).
the protein charges. We embedded the system in a
The most important point in the metadynamics is to
hydrophobic environment of detergent molecules (lauryl
select the proper reaction coordinates. These must be
dimethyl amine oxide, LDAO) and solvated the system
variables that are of interest but difficult to sample with a
with ∼8000 water molecules in an hexagonal box.
standard scheme, since the local minima in the space
Hexagonal periodic boundary conditions were used and
spanned by these variables are separated by barriers that
the simulation box edges are 68.4 Å, 68.4 Å, 78.1 Å.
cannot be overcome in the simulation time available. We
Electrostatic interactions at long distance were evaluated
also wanted to compare the effect of choosing different
using the soft particle mesh Ewald scheme while a cutoff of
collective variables in this case, and thus we chose the three
10 Å was used for the Lennard-Jones and short electrostatic
different collective variables:
energy terms. Multiple time step algorithm (MTS Respa)
(i) Distance (Z) , defined as: Z ¼ ZGEO an
ð tÞ� ZGEOðsoluteÞ,
was used with the SHAKE algorithm to keep bond lengthsinvolving hydrogens fixed. The simulations were done at
where ZGEO is calculated as the average of Z coordinates of
300 K with Nose thermostat to control temperature.
all the heavy atoms of the system, respectively antibiotics
We used the Amber potential for protein and TIP3P for
and (porin + detergent)
water []. The parameters of antibiotics were developed
(ii) Angle (Θ), defined as: CosðqÞ ¼ eZ:vant,
following the Amber protocol ]: (i) the three dimen-sional chemical structure of the antibiotic was drawn, using
with eZ being the eigenvector of inertia tensor (calculated
the software package HyperChem; (ii) geometry optimiza-
on the porin Cα) that is closest to the axe of diffusion of the
tion was performed using the Hartree-Fock (HF) basis set
porin, vant being the vector of the long axis of the antibiotic,
HF-6-31G* with the Gaussian03 package [(iii) the
the closest to the highest component of the dipole moment.
molecular electrostatic potential was generated at HF/6-
(iii) Number of hydrogen bonds, defined as a continuous
31G* level; (iv) the atomic charges were fitted to molecular
function from the list of all defined donors and
electrostatic potential with aid of a module RESP in Amber8
acceptors of the system:
program package, adding the restraint for equivalent atoms
HB ¼ i¼ 1�ðr Þ12
. The structure obtained after full optimization is
where nHB is the total number of possible hydrogen bonds
considered the starting geometry of the molecule onto
calculated at time t; r0 is the reference distance between the
which we assign all the force-field parameters. The atom
two heavy atoms of the bonds taken as 2.5 Å.
types and parameters were derived using either the program
The metadynamics algorithm enables the reconstruction
antechamber (Amber-module), when possible, or were
of the free energy in the subspace of the collective variables
assigned manually on the basis of our optimized geometry.
by integrating the history dependent terms Due to the
We used the program ORAC and the Amber force field [
complexity of the process studied, we calculated the free
energy after obtaining the first translocation path, which is
Further, we report the molecular properties in terms of
considered to be the most probable path because it passes
the amount of hydrophilic/hydrophobic surface, as calcu-
through the lowest saddle point, as done before for the
lated from the PLATINIUM server ].
unthreading of a molecule ]. In fact, once the antibioticcrosses the constriction region, we expected a diffusiveregime, with no significant affinity sites. The error bars
Results and discussion
associated with the energy calculations were assessed aspreviously done [and are of 1 kcal mol−1 at most.
Metadynamics simulations allow studyingthe translocation process
Microscopic analysis methodology
Both ampicillin and moxifloxacine have a permanentdipole, being its larger component along their long axis
To decipher the molecular details of the translocation
(Fig. b–c). As during permeation, antibiotics have to
mechanism additional equilibrium MD simulations (1 ns
penetrate the constriction region that is rich of charged
length) were started from each visited minima along the
amino acids, it is important to accurately define the way of
diffusion path. In depth analysis using VMD and in-house
entry and the interactions with the channel. To do so, we
scripts, was performed to characterize the following key
included explicitly these degrees of freedom as reaction
structural features:
coordinate along the metadynamics runs.
(i) The atomic root mean square fluctuations (rmsf) were
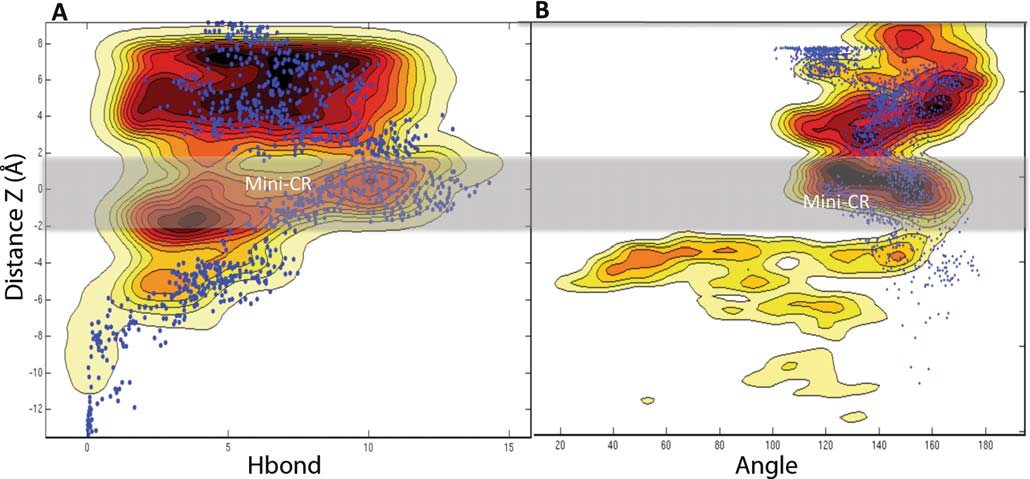
The Fig. displays the free energy surfaces (FES) of the
calculated for each heavy atoms of the backbone of the
translocation of the ampicillin beta-lactam antibiotic (Amp)
antibiotic with respect to its average position during the
through the OmpF channel for two different choices of the
reaction coordinates: (A) with the combination of the
(ii) Existence of hydrogen bonds (Hb) and hydrophobic
distance Z and the number of hydrogen bonds Hb; and
contacts (Hc) between atoms of the antibiotics and of
(B) with the combination of the distance Z and the angle Θ.
OmpF. Hbs are counted using VMD scripts according
Interestingly, the free energy barrier required for ampicillin
to the following threshold parameters: a distance of at
to translocates is of the same order in the two simulations:
most 3 Å and donor-hydrogen-acceptor angle of at
11 kcal mol−1 and 9 kcal mol−1 for the simulation A and B
least 130 degrees. Hcs are counted when non-polar
respectively (the values correspond to the energy barrier
atoms are separated by at most 3 Å.
needed to translocates starting from the deepest minima
Fig. 2 Complete free energy surfaces for ampicillin translocating
and the number of Hb; (B) the distance Z and the angle Θ. Each isoline
through OmpF, associated with the reaction pathway along the subspace
correspond to a different color gradient and to 1 kcal mol−1. The blue
of the two variables: taken as (A) the distance Z (Z=0 corresponds to
points superimposed on the FES correspond to the superimposed values
the center of the constriction zone, also indicated by the gray shading)
of the RC calculated from the other simulation

above). In both cases, deep energy minima are visited,
simulations for the translocation of moxiflocacine. First,
which can be related to well defined affinity sites of
the free energy barrier required for moxiflocacine to
ampicillin inside the channel. In particular, we observe the
translocate is much larger in the simulation A, 21 kcal
location of a deep energy minimum exactly at the
mol−1, compared to the simulation B, 16 kcal mol−1. The
constriction region (Mini-CR at Z∼0, see Fig. ). To
localization of the energy minima, defining the affinity sites
address the similarity (reproducibility) of the process
of the antibiotic inside the channel, is also very different. In
sampled with different RC, we further calculated the
the simulation A we note the absence of a deep energy
missing RC of each simulation. Thus, the Hb coordination
minimum at the constriction region, instead it is localized
number, as calculated in the course of the simulation B,
slightly above, at Z∼4 Å (Mini-Above, Fig. In the case
were superimposed as blue points in Fig. ; similarly, the
of the simulation B we find two energy minima localized
Θ angle, as calculated in the course of the simulation A,
above but also, in particular, one energy minimum is
were superimposed as blue points in Fig. Interestingly,
centrally localized at the constriction region, at Z∼0 Å
the number of hydrogen bonds needed (about 10 Hbs) to
cross the constriction region and translocate is similar in
The important differences between these simulations
both simulations that were "biased" (A) or "free" (B) for
using different RC are seen from the superimposition in the
this RC. Similarly, both simulations sample the same
FES of the calculated values of the missing RC. As seen in
populations of states of the angle Θ (the value of Θ when
the blue points superimposed in Fig. , the hydrogen
ampicillin crosses the constriction region and translocates is
bonds calculated from the simulation B do not follow the
around 150 degrees in both simulations A and B), such as
same path as the Hb sampled from simulation A. Instead,
in the deep minima at the constriction region. The fact that
the number of Hb calculated from the simulation B is both
both the angle and the hydrogen bond coordinate are
more constant and lower than the number of Hb sampled
reproducing a very similar path, with a deep energy minimum
along simulation A (∼4 Hb from simulation B while there
located centrally at the CR, comforts us in the choice of
are 8 Hb when the antibiotic crosses the constriction region
these RC. For the study of ampicillin's translocation, both
in simulation A). Similarly, as seen in Fig. , the
RC are appropriate.
superimposed Θ angles calculated from simulation A do
Next, we performed metadynamics simulations using the
not really follow the path of the Θ angles sampled in
same two sets of RC for the quinolone antibiotic: moxi-
simulation B. Instead, the Θ angles calculated from
floxacine. As seen in the FES displayed in Fig. , there are
simulation A reveal a much more stretched and limited
some important differences between the two sets of
exploration path (the Θ angles only take values from 40 to
Fig. 3 Complete free energy surfaces for moxifloxacine translocating through OmpF (see legends in Fig. )
120 degrees in the case of the simulation A, whereas they
explained in the Method section). We observe that
can cover fully the range from 0 to 180 in the case of
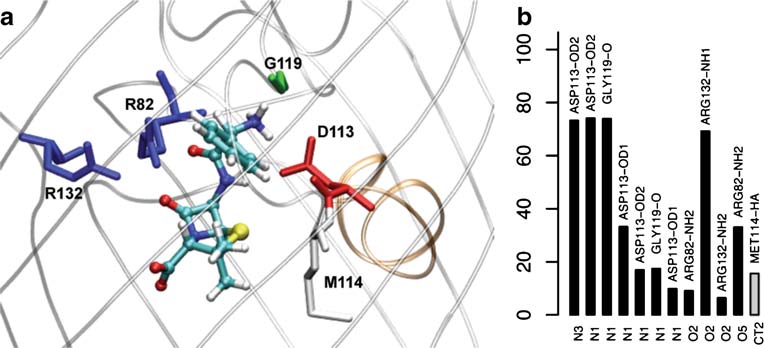
ampicillin is able to make a large number of durable polar
simulation B), as a consequence some local minima (above
Hbs, in particular both the one between its N-terminal
the constriction region) are not sampled in the path
positive group (N1) and D113 and the one between its C-
followed by the simulation A (Fig. ).
terminal carboxylic group (O2) and R132 exceed 70% of
To elucidate the microscopic details of the different
the equilibrium simulation time. Instead, only one signifi-
translocation path and conclude on the importance of the
cant Hc (∼20% of the simulation time) is made between the
choice of the RC we further performed in-depth analysis
CT2 carbon of ampicillin and M114 (Fig.
based on equilibrium simulations.
A similar detailed structural analysis was performed
when moxifloxacine is bound in Mini-CR, in the simulation
Equilibrium MD simulations allow studying the structural
B (with the Θ RC), and we find that in this case, the
and dynamics properties
antibiotic is oriented with its polar carboxyl group pointingup (Fig. ). Instead, it is with its hydrophobic group that
In order to characterize the structural and dynamics
moxifloxacine is seen to penetrate the constriction region
properties that govern the antibiotics diffusion process we
(Fig. ). The interaction network analysis indeed reveals a
then performed equilibrium MD simulations (of 1 ns
predominance of Hcontacts between the antibiotic and the
length) starting from the relevant, preferential minima,
many hydrophobic residues on the porin side such as F118,
identified by the FES. We focus only on the minima where
L20, M114, P116 (Fig. The only significant Hb
the antibiotics are bound at the constriction region of the
identified involves Y124, E117 and to a lesser extend R82.
channel (Mini-CR) as if such interaction was the rate-
Differences in the structural, dynamical or physico-
limiting step of the process. In the case of ampicillin, we
chemical properties of the two antibiotics could explain
find that very similar structures are sampled along the two
the differences observed in our study - that is, the translocation
Mini-CR from both simulations with the Hb and Θ RC, we
of ampicillin is described equivalently by both the Hb and the
will further only describe the Mini-CR of the simulation B
Θ RC while the translocation process of moxifloxacine differs
(with the Θ RC). As seen from the snapshot in Fig. , to
if choosing the Hb or the Θ RC. Table reports diverse
enter the affinity site at the constriction region (Mini-CR)
physico-chemical properties of the two antibiotics, such as
and further translocates, ampicillin is oriented with its polar
the flexibility and solvation pattern, obtained from the
carboxyl group pointing down and makes favorable
optimized 3D structures or from simulations where they are
interactions with the residues of the porin constriction
placed in a box of water. We observe that, although both
zone. We present in Fig. the lifetime of the hydrogen
antibiotics have the same molecular surface size, ampicillin
bonds and hydrophobic contact (Hc) interactions between
is found to be much more polar while moxifloxacine is much
ampicillin and OmpF atoms sampled along Mini-CR (as
more hydrophobic (see computed surface properties in
Fig. 4 a) Molecular detail of ampicillin at the preferred minimum, at
the constriction region (Mini-CR). In the x-axis is represented each
the constriction region, Mini-CR, showing the interacting residues of
atom of the antibiotic, in y-axis is calculated the lifetime (probability
OmpF (colored by residue types, the ones involved in Hbs are
of existence along simulation) of each interaction between atoms of
displayed as sticks and those involved in Hcontacts are displayed by
the antibiotic and atoms of OmpF. In black bars are represented the
molecular surface) (see Fig. for coloring code). b) Interaction map
hydrogen bonds (Hb) and in gray bars the hydrophobic contacts
for the equilibrium MD of ampicillin in the preferential minimum, at
(Hcontacts), calculated as explained in
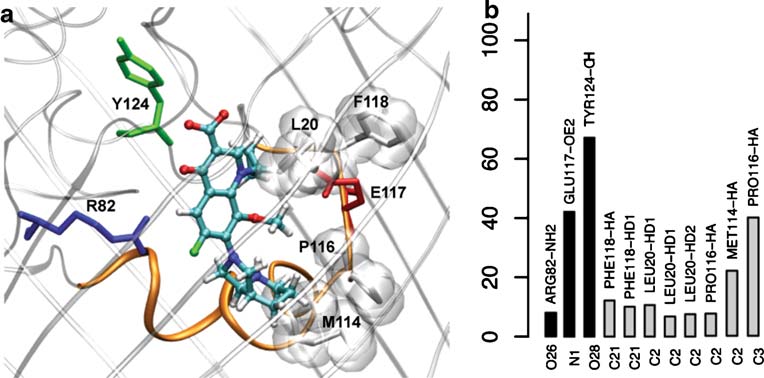
Fig. 5 Molecular details (a) andinteraction map (b) of moxi-floxacine at the preferentialminimum, Mini-CR (see legendsin Fig. )
Table ). This is in agreement with the antibiotics nature of
Altogether, from our simulations, we observe that the
interactions with OmpF. Interestingly, we also find that the
bottleneck for the antibiotics translocation is to overcome
ampicillin structure is twice more flexible than the one of
the constriction region, where the antibiotic has to
moxifloxacine (see calculated rmsf in Table
optimally interact with the channel, its dipole has to matchthe strong transversal electrostatic field and its surface hasto adapt to fit to a given size.
In the simulation where the Hb was taken as a RC, we
found that ampicillin can make and break Hb easily and it
Here we used all-atom metadynamics simulations with a
reaches up to 10 Hb but in the case of moxifloxacine this
different choice of the RC and followed the translocation
number is constant and as low as 4, thus we believe that
through OmpF of two commonly used antibiotics. The high
overcoming the constriction is more difficult in the case of
resolution in time and space that metadynamics simulations
a less polar or more hydrophobic antibiotic. The molecular
provide has the potential to be used to characterize the
details of the simulations allow assessing how the choice of
molecular basis of translocation of antibiotics, and further,
the RC affects the FES and the molecular mechanism for
such strategy might be used to help select/design antibiotics
with better permeation properties and thus combat bacterial
In the case of ampicillin, although metadynamics is
resistance. To improve the accuracy and efficiency of future
applied with a different RC, we find a similar antibiotic
simulations studies, we must keep on developing the algo-
translocation pathway. In both cases, a unique energy
rithms at hands. Here we address the consequences of choosing
minimum is found for ampicillin at the constriction region,
a different RC, which is the crucial point of the accelerated
which is well defined with a strong interaction network. In
molecular dynamics techniques. Due to the cylindrical shape of
this preferential minimum, the (two charged groups) dipole
OmpF, a tentative choice of RC is the position of the
of the antibiotic matches perfectly with the one of the porin,
antibiotic along the main axe of diffusion. Our results on
known as the transversal electric field, with negative
moxifloxacin and ampicillin support our hypothesis that the
residues clustered on the L3 side and positive residues
Θ angle defines an optimal RC, as it is an internal variable
lining on the anti-L3 side (see Fig.
not subject to change as OmpF diffuses during simulation.
Interestingly, a different molecular path is found by
moxifloxacine with respect to ampicillin: it translocateswith its hydrophobic group pointing down and we note a
Table 1 Structural details of the antibiotic obtained from the
predominance of Hcontacts interactions that play an
equilibrium MD simulations (see )
important contribution as a driving force. Such a possiblecontribution from hydrophobicity was already raised in the
Surface properties []
(in box of waters)
pioneer work of Nikaido ]. Indeed, the newer gener-
ations of antibiotics that are more hydrophilic have
enhanced antibacterial activity, indicating that hydrophilic-ity would enhance their affinity to OmpF. Thus, in such
cases, we suggest to include directly the hydrophobic
interactions number as a RC in the course of metadynamics
simulations. Further analysis allowed extracting important
4. Nestorovich EM, Danelon C, Winterhalter M, Bezrukov SM
structural and dynamical properties of the antibiotics, such
(2002) Designed to penetrate: time-resolved interaction of singleantibiotic molecules with bacterial pores. Proc Natl Acad Sci USA
as solvation and flexibility, and the same could be done
with the porin target.
5. Ceccarelli M, Danelon C, Laio A, Parrinello M (2004) Micro-
It is noteworthy that the two different sets of reaction
scopic mechanism of antibiotics translocation through a porin.
coordinates used in this manuscript reproduce well the
Biophys J 87:58–64
6. Laio A, Parrinello M (2002) Escaping free-energy minima. Proc
previously obtained experimental observations [].
Natl Acad Sci USA 99:12562–12566
Indeed, both resulting simulations reveal the same "macro-
7. Robertson KM, Tieleman DP (2002) Orientation and interactions
scopic event" that is the passage/diffusion of the antibiotic
of dipolar molecules during transport through OmpF porin. FEBS
and both find a preferential affinity site for the antibiotic
8. Henin J, Tajkhorshid E, Schulten K, Chipot C (2008) Diffusion of
inside the channel. However, using different reaction
glycerol through Escherichia coli aquaglyceroporin GlpF. Biophys
coordinates leads to different simulation time and efficien-
cy. In this particular manuscript we aim at improving the
9. Vidal S, Bredin J, Pages JM, Barbe J (2005) Beta-lactam
efficiency of the molecular simulations and we show how
screening by specific residues of the OmpF eyelet. J Med Chem48:1395–1400
the "right choice of coordinate" could be primordial for
10. Mach T, Neves P, Spiga E, Weingart H, Winterhalter M,
doing so. The importance of having an efficient and reliable
Ruggerone P, Ceccarelli M, Gameiro P (2008) Facilitated
computational model for antibiotic translocation is also
permeation of antibiotics across membrane channels-interaction
highlighted by our recent findings that pointed out the
of the quinolone moxifloxacin with the OmpF channel. J AmChem Soc 130:13301–13309
limitations, in time and space resolutions, of kinetic
11. Danelon C, Nestorovich EM, Winterhalter M, Ceccarelli M,
Bezrukov SM (2006) Interaction of zwitterionic penicillins with
To conclude we believe that to gain in efficiency and
the OmpF channel facilitates their translocation. Biophys J
accuracy, the antibiotic translocation could be simulated
12. Im W, Roux B (2002) Ions and counterions in a biological
with many RC coupled together: allowing to account
channel: a molecular dynamics simulation of OmpF porin from
explicitly for solvation, flexibility and different possible
Escherichia coli in an explicit membrane with 1 M KCl aqueous
specific interactions/properties of importance in the system
salt solution. J Mol Biol 319:1177
of study. This could be done by using a scheme such as the
13. Cornell WD et al. (1995) J Am Chem Soc 117:5179
14. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004)
bias exchange metadynamics" recently proposed [
Development and testing of a general amber force field. J Comput
Another possibility is to use a bias-free method [but
Chem 25:1157–1174
then a way must be found to sample the trajectory
15. Frisch MJ et al. (2004) Gaussian 03, Revision C02. Gaussian Inc,
corresponding to the studied process (for example, by
16. Procacci P, Darden TA, Paci E, Marchi M (1997) ORAC: a
using temperature as a bias as in TAMD simulations ]).
molecular dynamics program to simulate complex molecular
Such explorative strategies would in fact reveal which RC
systems with realistic electrostatic interactions. J Comput Chem
is relevant for the process and system studied. Better-tuned
simulation methodology will also be useful to investigate
17. Laio A, Rodriguez-Fortea A, Gervasio FL, Ceccarelli M,
Parrinello M (2005) Assessing the accuracy of metadynamics.
many diverse complex biological processes, such as folding
J Phys Chem B 109:6714–6721
or protein-protein interactions.
18. Pyrkov TV, Chugunov AO, Krylov NA, Nolde DE, Efremov RG
(2009) PLATINUM: a web tool for analysis of hydrophobic/
We thank Attilio Vargiu and Enrico Spiga for
hydrophilic organization of biomolecular complexes. Bioinfor-
fruitful discussions. This study was supported by EU-grant MRTN-
matics 25:1201–1202
CT-2005-019335 (Translocation) and by the computer center and
19. Yoshimura F, Nikaido H (1985) Diffusion of beta-lactam anti-
consortiums: Cybersar, CASPUR and CINECA through CPU-hours.
biotics through the porin channels of Escherichia coli K-12.
Antimicrob Agents Chemother 27:84–92
20. Hajjar E, Mahendran KR, Kumar A, Bessonov A, Petrescu M,
Weingart H, Ruggerone P, Winterhalter M, Ceccarelli M (2010)
Bridging timescales and length scales: from macroscopic flux tothe molecular mechanism of antibiotic diffusion through porins.
1. Nikaido H (2003) Molecular basis of bacterial outer membrane
Biophys J 98:569–575
permeability revisited. Microbiol Mol Biol Rev 67:593–656
21. Piana S, Laio A (2007) A bias-exchange approach to protein
2. Chevalier J, Mallea M, Pages JM (2000) Comparative aspects of
folding. J Phys Chem B 111:4553–4559
the diffusion of norfloxacin, cefepime and spermine through the F
22. Branduardi D, Gervasio FL, Parrinello M (2007) From A to B in
porin channel of Enterobacter cloacae. Biochem J 348:223–227
free energy space. J Chem Phys 126:54103
3. Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit
23. Weinan E, Vanden-Eijnden E (2010) Transition-path theory and
RA, Jansonius JN, Rosenbusch JP (1992) Crystal structures explain
path-finding algorithms for the study of rare events. Annu Rev
functional properties of two E. coli porins. Nature 358:727–733
Source: http://dottorato.dsf.unica.it/pdf/pubblicazioni/44-2010.pdf
Department of Health and Human Services OFFICE OF INSPECTOR GENERAL PART D BENEFICIARIES WITH QUESTIONABLE UTILIZATION PATTERNS FOR HIV DRUGS Daniel R. Levinson Inspector General August 2014 EXECUTIVE SUMMARY: Part D Beneficiaries With Questionable Utilization Patterns for HIV Drugs, OEI-02-11-00170
Translating cell biology intotherapeutic advancesin Alzheimer's diseaseDennis J. Selkoe Studies of the molecular basis of Alzheimer's disease exemplify the increasingly blurred distinction between basic andapplied biomedical research. The four genes so far implicated in familial Alzheimer's disease have each been shown toelevate brain levels of the self-aggregating amyloid-b protein, leading gradually to profound neuronal and glialalteration, synaptic loss and dementia. Progress in understanding this cascade has helped to identify specifictherapeutic targets and provides a model for elucidating other neurodegenerative disorders.



