Viagra gibt es mittlerweile nicht nur als Original, sondern auch in Form von Generika. Diese enthalten denselben Wirkstoff Sildenafil. Patienten suchen deshalb nach viagra generika schweiz, um ein günstigeres Präparat zu finden. Unterschiede bestehen oft nur in Verpackung und Preis.
Stressresponse balance drives the evolution of a network module and its host genome
Published online: August 31, 2015
Stress-response balance drives the evolution of anetwork module and its host genome
Caleb González1,†, Joe Christian J Ray1,2,†, Michael Manhart3,4, Rhys M Adams1, Dmitry Nevozhay1,5,
Alexandre V Morozov3,6 & Gábor Balázsi1,7,8,*
have expanded quickly, feeding on general biological knowledge.
Conversely, synthetic biology has enormous but unexploited poten-
Stress response genes and their regulators form networks that
tial to inform other areas of biology, such as evolutionary biology
underlie drug resistance. These networks often have an inherent
(Tanouchi et al, 2012b).
tradeoff: their expression is costly in the absence of stress, but
For example, gene regulatory networks that control the expression
beneficial in stress. They can quickly emerge in the genomes of
of stress-protective genes have emerged through evolution (Lopez-
infectious microbes and cancer cells, protecting them from treat-
Maury et al, 2008) but can also be built de novo (Nevozhay et al,
ment. Yet, the evolution of stress resistance networks is not well
2012; Tanouchi et al, 2012a). Depending on the details of gene regu-
understood. Here, we use a two-component synthetic gene circuit
lation, cells can survive because they respond to stress (Gasch et al,
integrated into the budding yeast genome to model experimentally
2000); diversify non-genetically (hedge bets), independent of the
the adaptation of a stress response module and its host genome in
stress (Balaban et al, 2004; Thattai & van Oudenaarden, 2004; Levy
three different scenarios. In agreement with computational predic-
et al, 2012); or use a mixture of these two strategies (New et al,
tions, we find that: (i) intra-module mutations target and elimi-
2014). However, stress-protective gene expression can be costly or
nate the module if it confers only cost without any benefit to the
toxic in the absence of stress (Andersson & Levin, 1999), or even in
cell; (ii) intra- and extra-module mutations jointly activate the
the presence of stress when the expression level exceeds the require-
module if it is potentially beneficial and confers no cost; and (iii) a
ment for survival (Nevozhay et al, 2012). Overall, the costs and bene-
few specific mutations repeatedly fine-tune the module's noisy
fits of survival mechanisms create a tradeoff between maximizing
response if it has excessive costs and/or insufficient benefits. Over-
growth while also ensuring survival during stress. How mutations
all, these findings reveal how the timing and mechanisms of stress
alter stress response networks to improve fitness under such circum-
response network evolution depend on the environment.
stances, especially in phenotypically heterogeneous populations(Sumner & Avery, 2002), is an open problem in evolutionary biology.
Keywords drug resistance; experimental evolution; positive feedback;
Consider a stress response network module, consisting of a
synthetic gene circuit; tradeoff
stress-sensing transcriptional regulator and its stress-protective gene
Subject Categories Quantitative Biology & Dynamical Systems; Synthetic
target, which has arisen in a cell's genome. Similar modules, such
Biology & Biotechnology; Evolution
as Tn10 (Hillen & Berens, 1994), toxin-antitoxin systems (Yama-
DOI 10.15252/msb.20156185 Received 21 March 2015 Revised 31 July 2015
guchi et al, 2011), or bypass signaling (Hsieh & Moasser, 2007), can
Accepted 4 June 2015
arise rapidly by recombination, horizontal gene transfer, or inhi-
Mol Syst Biol. (2015) 11: 827
bitor-mediated alternate pathway activation. Considering theirimpact on microbial and cancer drug resistance, it is important toknow how reproducibly and how quickly such stress defense
networks can adapt (Lobkovsky & Koonin, 2012). Yet, we currentlylack quantitative, hypothesis-driven understanding of how initially
The number of human-designed biological systems has increased
suboptimal stress defense modules evolve inside the host genome,
rapidly since the inception of synthetic biology (Purnick & Weiss,
especially in the presence of gene expression noise (Bala´zsi et al,
2009). Parts and concepts underlying synthetic biological constructs
2011; Munsky et al, 2012; Sanchez & Golding, 2013). Although
1 Department of Systems Biology - Unit 950, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
2 Center for Computational Biology & Department of Molecular Biosciences, University of Kansas, Lawrence, KS, USA
3 Department of Physics & Astronomy, Rutgers University, Piscataway, NJ, USA
4 Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA, USA
5 School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia
6 BioMaPS Institute for Quantitative Biology, Rutgers University, Piscataway, NJ, USA
7 Laufer Center for Physical & Quantitative Biology, Stony Brook University, Stony Brook, NY, USA
8 Department of Biomedical Engineering, Stony Brook University, Stony Brook, NY, USA
*Corresponding author. Tel: +1 631 632 5414; Fax: +1 631 632 5405; E-mail:
[email protected]
†These authors contributed equally to this study
ª 2015 The Authors. Published under the terms of the CC BY 4.0 license
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
network evolution theory (Kauffman, 1993; Mason et al, 2004;
what mechanisms drug resistance emerges or deteriorates in the
Kashtan & Alon, 2005) and laboratory evolution experiments
process of network evolution, and could help the future design of
(Lenski & Travisano, 1994; Beaumont et al, 2009; Tenaillon et al,
synthetic gene circuits that resist evolutionary degradation.
2012; Toprak et al, 2012; Lang et al, 2013) have generated impor-tant insights, they have provided largely descriptive, a posterioriinterpretations. Now there is a growing need for predictive, hypoth-
esis-driven, quantitative understanding of gene network evolution,which requires making a priori predictions of mutation effects and
The PF gene circuit can mimic various scenarios of stress-
evolutionary dynamics that are tested experimentally (Wang et al,
response imbalance
2013). One option could be to study the evolution of small naturalregulatory modules (Dekel & Alon, 2005; Hsu et al, 2012; Quan
We considered the following disparities between the external stress
et al, 2012; van Ditmarsch et al, 2013). However, connections of
and the activity of a stress defense module: (i) the module responds
natural regulatory modules with the rest of the genome can be
gratuitously to a harmless environmental change; (ii) the module
significant (Maynard et al, 2010) and poorly characterized, thus
cannot respond to harmful stress when needed; and (iii) the module
making predictive, quantitative understanding difficult. Synthetic
responds to stress, but suboptimally. To mimic these scenarios
gene circuits (Elowitz & Leibler, 2000; Gardner et al, 2000; Stricker
using the PF gene circuit in yeast, we relied on the separability of
et al, 2008; Moon et al, 2012; Nevozhay et al, 2013) represent a
stress and response, adjusting two environmental factors with
better alternative, since they are small, consist of well-characterized
known fitness effects (Nevozhay et al, 2012): inducer doxycycline
components, and typically lack direct regulatory interactions with
and antibiotic zeocin (Fig 1). Hereafter, DxZy will denote environ-
the host genome. However, it is unclear whether the evolution of
mental conditions, with x and y indicating doxycycline and zeocin
synthetic gene circuits (Yokobayashi et al, 2002; Sleight et al, 2010;
concentrations, respectively. The antibacterial compound doxycy-
Poelwijk et al, 2011; Wu et al, 2014) can be predicted a priori,
cline has negligible effect on yeast (Wishart et al, 2005), but causes
especially with regard to gene expression heterogeneity.
squelching toxicity in engineered PF cells when bound to rtTA (Gari
We recently characterized the dynamics and fitness effects of
et al, 1997; Nevozhay et al, 2012). Zeocin is a broad-spectrum
gene expression for a synthetic two-gene "positive feedback" (PF)
DNA-damaging antibiotic (Burger, 1998) that acts on bacteria and
circuit (Fig 1A) integrated into the genome of the haploid single-
celled eukaryote Saccharomyces cerevisiae (Nevozhay et al, 2012).
First, the presence of inducer doxycycline alone corresponds to
This synthetic gene circuit consists of a well-characterized transcrip-
scenario (i): costly, futile response of some (Fig 1B, DiZ0) or most
tional regulator (rtTA) and an antibiotic resistance gene (yEGFP::
(Fig 1B, D2Z0) cells that start expressing the PF genes. The cost of
zeoR). In the presence of tetracycline-analog inducers such as doxy-
response slows the cell division rate of responding, high expressor
cycline, rtTA activates both itself and yEGFP::zeoR by binding to two
cells compared to non-responding, low expressor cells (Nevozhay
tetO2 operator sites in two identical promoters (Fig 1A). This posi-
et al, 2012). Consequently, the division rate of individual yeast
tive feedback is noisy, however, and thus, only a fraction of cells
cells can differ drastically from the overall population growth rate.
switch to high expression of rtTA and yEGFP::zeoR. These cells
To capture these differences between single cell- and population
benefit from high gene expression, which protects them from the
growth rates, we constructed a population fitness landscape (three-
antibiotic zeocin. Meanwhile, the same cells experience a cost from
dimensional gray surface in Fig 1B) and cellular fitness landscapes
rtTA activator expression toxicity, causing a tradeoff when zeocin is
(colored panels in Fig 1B). The population fitness landscape maps
present (Nevozhay et al, 2012). The fitness (division rate) of any
the overall population growth against the two environmental vari-
individual cell is the product of its rtTA expression cost and yEGFP::
ables, doxycycline and zeocin concentrations. Cellular fitness land-
zeoR expression benefit (Nevozhay et al, 2012), which varies from
scapes depict the division rate of single cells versus their gene
cell to cell. Thus, quantitative knowledge of dynamics and fitness
expression level in a given combination of doxycycline and zeocin.
effects makes the PF gene circuit an excellent model for studying
As described in the Appendix, we inferred these landscapes
gene network evolution in tradeoff situations. Its design separates
directly from growth rate and gene expression measurements
stress (zeocin) from its adjustable cellular response (inducible
(Appendix Fig S1A) in 13 different combinations of doxycycline
yEGFP::zeoR expression), facilitating predictive, quantitative under-
standing of how a stress response module adapts inside the host
Second, the presence of antibiotic zeocin alone (Fig 1B, D0Z2)
corresponds to the lack of response when needed, as in scenario (ii).
Here, we used our quantitative knowledge of the PF gene circuit
Finally, the presence of both inducer and antibiotic (Fig 1B, DiZ2 and
to predict a priori the timing and mechanisms of its initial adaptation
D2Z2) corresponds to scenario (iii) where the fraction of responding,
to several constant environments (squares in Fig 1B) corresponding to
slower-growing cells ensures cell population survival during antibiotic
various stress-response imbalance scenarios. We tested these
treatment, but the response is in general suboptimal.
predictions with experimental evolution, followed by sequencing to
Altogether, the PF gene circuit is a well-characterized module
identify the mutations that establish in the population, depending
lacking direct regulatory interactions with the yeast genome. It
on the imbalance between the environmental stress and the intracel-
exemplifies typical tradeoffs between the benefits and costs of gene
lular response. In this way, we tested how different mutations can
expression in stress response networks. Importantly, the benefits
readjust the response of a network module with inherent tradeoff,
and costs are independently tunable for the PF gene circuit,
to match the stress and minimize the cost in each specific environ-
making it possible to predict and test their evolution toward
ment. These results could help us understand how fast and through
Molecular Systems Biology
ª 2015 The Authors

Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
F F F
D0Z0 (flat)
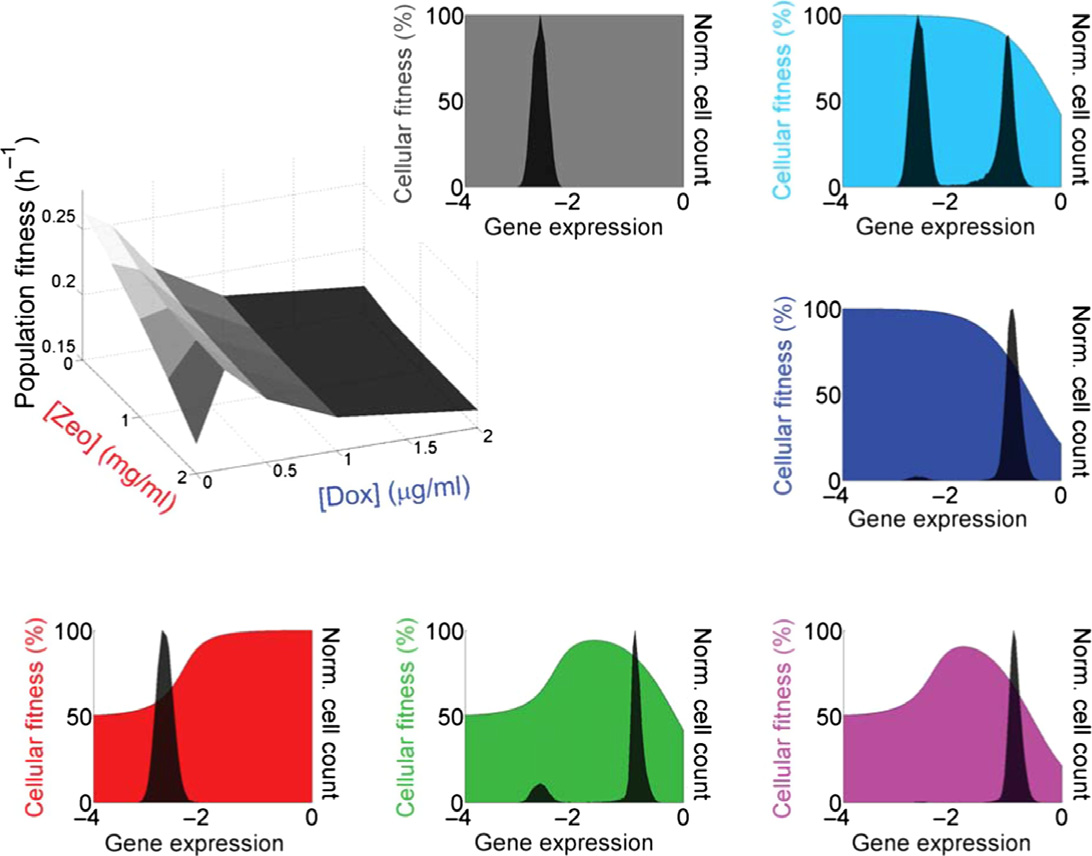
Figure 1. The PF synthetic gene circuit: fitness and gene expression characteristics.
A The PF synthetic gene circuit (Nevozhay et al, 2012) consists of two components. First, the regulator reverse tet-trans-activator (rtTA) (Urlinger et al, 2000) is a
reverse-tetR gene fused to three F activator domains (cyan rectangles), which are shorter versions of the VP16 activator (Baron et al, 1997). The target geneyEGFP∷ZeoR consists of the fluorescent reporter yEGFP fused to the drug resistance gene zeoR (Gatignol et al, 1988) that binds and inactivates zeocin, a bleomycin-family antibiotic. Unbound zeocin generates DNA double-strand breaks, causing cell cycle arrest and potentially cell death. Doxycycline added to the growth mediumdiffuses freely through the cell wall and binds to rtTA dimers. Inducer-bound rtTA undergoes a conformational change that results in strong association with twotetO2 operator sites upstream of each of the two tetreg promoters (Becskei et al, 2001), activating both regulator and target gene expression, while causing toxicityby squelching.
B Costs and benefits of PF gene circuit components were determined by measuring cell population growth rate (population fitness) versus two environmental factors:
inducer doxycycline and antibiotic zeocin. Each point on the population fitness landscape (three-dimensional gray surface on the left) is an average of cellular fitnessvalues (color-shaded slopes in the surrounding plots) as cells stochastically move within gene expression distributions (black histograms in the surrounding plots).
Gene expression is measured as log10(fluorescence) (arbitrary units). DxZy denotes the environment (the x and y following D and Z indicate lg/ml doxycycline andmg/ml zeocin concentrations, respectively, with Di = 0.2 lg/ml doxycycline). Cellular fitness (cell division rate) is a function of gene expression for each cell in eachenvironment DxZy. It is inferred from the population fitness, based on a biochemical model (Nevozhay et al, 2012); see the Appendix. The black arrows beneathcellular fitness landscapes illustrate selection pressures pushing the gene expression distribution toward higher fitness.
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
Predicting the first evolutionary steps in constant environments
randomly to a level corresponding to intermediate expression on thecellular fitness landscapes. Finally, G-type mutants increased their
We asked whether the PF cellular and population fitness land-
fitness randomly, up to a level they would have without zeocin.
scapes (colored squares and panels in Fig 1B; Appendix Fig S1A;
This simpler model could predict how fast the wild-type genotype
Appendix Table S1) could predict evolutionary trends in specific
disappears from the population. It could also forecast the mutation
environments. For example, in the D2Z0 environment, most cells
type (K, T, G) that predominantly replaces the wild type in each
are far from their fitness maximum, which is at low expression. If
condition. However, it could not predict the number of distinct
a mutation could push cells downward in expression, toward their
mutant alleles in the evolving population. Moreover, it lacked
fitness maximum (horizontal arrow in Fig 1B, D2Z0 panel), then
potentially important experimental details, such as periodic resus-
they should grow faster. Mutations that either abolish or weaken
pensions and phenotypic switching.
rtTA toxicity could achieve this effect. Let us call these mutation
To test the importance of such additional details, the detailed
types "knockout" (K) and "tweaking" (T) mutations, respectively
simulation framework captured multiple experimentally relevant
(Fig 2A; Appendix Fig S1B). On the other hand, in the D0Z2 envi-
aspects of evolution. For example, cells could switch between On
ronment cells should benefit from mutations that diminish the
and Off states with experimentally inferred rates (Appendix
effect of the antibiotic. This could happen in various ways, for
Table S1). K, T, and G mutations with altered switching and growth
example by upregulation of native stress-response mechanisms; or
rates entered the population as single cells at a constant, but adjus-
by increasing yEGFP::zeoR expression. Let us call these latter
table rate l per cell per generation (Fig 2A; Appendix Fig S1C).
mutation types "generic" (G) drug resistance mutations (Fig 2A;
K-type mutants could not switch On, and thus had no cellular fitness
Appendix Fig S1B). In all these cases, mutant cells can improve
costs in doxycycline. T-type mutants switched On at a randomly
their fitness by unidirectionally lowering or increasing PF gene
reduced rate, and thus had diminished cellular fitness costs from PF
expression. However, in certain conditions (such as DiZ2), when
gene expression. G-type mutant cells had randomly increased drug
the cells form two subpopulations that flank the cellular fitness
resistance without any change in switching rates. We simulated
peak, a single-directional expression change is not optimal. This is
periodic resuspensions by repeatedly reducing the cell population
because a one-way expression shift can only move one subpopula-
size to 106. We considered cells to be initially drug- and inducer-
tion toward the fitness peak, while the other subpopulation must
free, and allowed them to gradually take up zeocin and doxycycline.
necessarily move away from it. Instead, optimally the two sub-
This simulation framework could predict the number of distinct
populations should approach each other, both moving toward the
mutant alleles, in addition to the characteristics predicted by the
fitness peak (horizontal arrows in Fig 1B, DiZ2, D2Z2 panels).
simpler model.
How would the PF cells evolve to adapt in specific combinations
Both models had three free parameters: the rate of potentially
of doxycycline and zeocin? Mutations of any type (K, T, G) can arise
beneficial mutations l, and the input probabilities P(G) and P(T) of
spontaneously, then establish in the population, and compete with
a given mutation being of type G or T, respectively. Once known,
each other depending on two requirements. First, the mutation type
these parameters also define the probability of a mutation to be of
must be available (genetic changes causing the phenotype must
type K: P(K) = 1 – P(G) – P(T). We note the difference between the
exist). Second, since we consider large populations, the mutation
rate and probability of a mutation: for example, the probability of
should be beneficial, improving fitness in the given environment.
P(K) could be equal to 1, while its rate lP(K) is much < 1 per
Despite these intuitive expectations, it is unclear how many muta-
genome per generation. Figure 2A depicts the effect of each muta-
tions of each type will establish in each condition, and how fast.
tion type, illustrating the relationships among the free parameters.
To address these questions in silico, we developed two comple-
We extracted the rest of the parameters (Appendix Table S1) from
mentary modeling approaches: a simple mathematical model and a
experimental measurements (see the Appendix) and kept them
detailed computational simulation framework (see the Computa-
tional Models.zip file and the Appendix for detailed descriptions).
Using these models, we studied how the three free parameters
The two models serve to test the robustness of results to various
affected three features of evolutionary dynamics: the ancestral geno-
modeling approaches. The simple model was more general and
type's half-life, as well as the type and number of mutant alleles in
faster, allowing more extensive parameter scans. On the other hand,
each condition (Fig 2; Appendix Figs S2 and S3). We started by
the simulation framework allowed testing how specific details of
studying the ancestral genotype's half-life in each model, scanning
experimental evolution would affect the evolutionary dynamics, and
each free parameter systematically (Fig 2B; Appendix Figs S2B and
provided more detailed results. We initiated both models with a
S3B). The models consistently indicated (Fig 2B) that the ancestral
population of ancestral (wild-type) PF cells, aiming to find out the
genotype disappeared fastest in conditions with steep monotone
number and type of mutations that establish and when the ancestral
cellular fitness landscapes (Fig 1B, D0Z2 and D2Z0). In contrast,
genotype disappears. We modeled 20 days of evolution in each
the ancestral genotype remained in the population longer in peaked
environment indicated by the colored squares in Fig 1B.
cellular fitness landscapes (Fig 1B, D2Z2 and DiZ2). Finally, the
The simpler model described population dynamics by a system
majority of cells were still genetically ancestral after 20 days in
of ordinary differential equations (ODEs), assuming constant popu-
DiZ0, which has the most gradual cellular fitness landscape (Fig 1B,
lation size and mutation rate. We characterized wild-type and
DiZ0). The time when the ancestral genotype disappeared in
mutant cells by a single parameter: their fitness (exponential growth
various environments depended differently on the mutation proba-
rate), determined from the fitness landscapes in Fig 1B. For exam-
bilities P(K), P(T), P(G) (Appendix Fig S3B). For example, the
ple, we assumed that K mutants had cellular fitness corresponding
ancestral genotype disappeared later in D2Z2 when we lowered P
to null expression in Fig 1B. T-type mutant cells altered their fitness
(T). Likewise, lowering P(G) prolonged the ancestral genotype's
Molecular Systems Biology
ª 2015 The Authors
Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
Relative Probability of T Mutations
Relative Probability of T Mutations
Fraction of Mutants
Figure 2. Simulation framework predicts evolutionary dynamics.
Simulating the initial steps of evolution. Three types of potentially beneficial mutations (with an overall rate l) enter the ancestral population of yeast cells thatinitially carry the intact PF gene circuit. Each cell can divide and mutate, producing new genotypes with altered fitness that can belong to three different types. Thefirst two types are knockout (K) and tweaking (T) mutations. They eliminate rtTA's regulator activity and toxicity completely or partially, respectively. The third typeincludes extra-rtTA or generic (G) mutations that cause zeocin resistance independently of rtTA. In the models, we consider exponential growth with randomelimination of cells or periodic resuspensions to control population size. Empty circles represent intact PF cells, while blue, magenta, and orange circles represent K, T,and G mutants, respectively. These mutations can arise, be lost, or expand in the population.
The speed at which mutants take over the population in each simulated condition is measured as the ancestral genotype's half-life (the time until only 50% of thepopulation carries the ancestral genome). N = 100; mean � SEM in each simulated condition: D2Z0, DiZ0, D0Z2, D2Z2, and DiZ2. In these plots, we fixedl�Z = 10�6.2 or l+Z = 10�5.4/genome/generation (for no zeocin and zeocin, respectively) and P(G) = 0.75. Therefore, P(T) = 0.25 � P(K). On the horizontal axis, weshow the probability of T mutations among intra-rtTA mutations: P*(T) = P(T ¬G), which scales P(T) four-fold up such that its maximum is 1 instead of 0.25. The graybar denotes the value used for time course simulations in subsequent figures. The parameter set for the gray bar on this and the following panels is P(T) = 0.025;P(K) = 0.225; and P(G) = 0.75.
C Number of established mutations with frequency > 5% at day 20. N = 100; mean � SEM in each simulated condition: D2Z0, DiZ0, D0Z2, D2Z2, and DiZ2. Parameters,
axes, and gray bar: as in (B).
D Population fractions of T-, K-, and G-type mutations at day 20, for the parameters corresponding to the gray bar, as indicated above.
presence in the populations in D0Z2. These observations confirmed
quantitative estimates of the speeds at which mutants establish and
the expectation that the most beneficial mutation in each condition
take over the evolving population.
dictates evolutionary dynamics. Overall, we hypothesized based on
In general, K, T, and G allele frequencies at the end of simulated
these results that the ancestral PF gene circuit should disappear
time courses did not match the input probabilities of P(K), P(T),
fastest in D2Z0 and D0Z2, followed by DiZ2 and D2Z2, and finally
and P(G) mutations. Rather, each condition favored different muta-
in DiZ0. Making these predictions required quantitatively under-
tion types as long as they were available (Fig 2D; Appendix Figs S2
standing the fitness properties and genetic structure of the PF gene
and S3). For example, in D2Z0, nearly all mutations were K-type
circuit. Without modeling, it would have been impossible to obtain
even if K mutations were unlikely to enter the population. T
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
mutations established exclusively in D2Z2, while in DiZ2 they
multiple days, we collected samples for whole-genome and tradi-
appeared alongside G mutations. In DiZ0, K or T mutations estab-
tional (Sanger) sequencing to reveal the mutations underlying the
lished late and spread slowly, with parameter-dependent relative
observed gene expression changes.
fractions. Finally, only G alleles could establish in D0Z2. Toconclude, both models predicted the environment-specific domi-
Scenario (i): reproducible circuit failure from gratuitous
nance of various mutation types at 20 days, irrespective of the
relative supplies of different mutation types. The most likely causeis each condition selecting one mutation type so strongly that the
To test the fate of a new stress defense module that becomes costly
final outcome of evolution (but not its dynamics) becomes quasi-
by gratuitously responding to an otherwise harmless environmental
deterministic. The long-term dominance of specific mutants in each
change, we grew PF yeast cells in inducer doxycycline without
condition might have been intuitively inferable from the fitness
antibiotic (D2Z0), resuspending every 12 h. In this condition, fluo-
properties and genetic structure of the PF gene circuit. However,
rescence first rose and then began to decline toward the basal level
modeling is indispensable to understand the evolutionary dynamics
in < 1 week ( 40 generations) for all three replicate populations
of mutants arising, establishing and competing before reaching the
(Fig 3A). The fluorescence decline continued until gene expression
final state.
was indistinguishable from that of uninduced cells by the end of the
Finally, we used the simulation framework to determine the
experiment, consistent with the effect of K-type mutations. As fluo-
number of alleles over 20 days in each condition (Fig 2C). This is
rescence levels dropped, population growth rate increased signifi-
perhaps the least intuitive result that could not have been predicted
cantly (see the Source Data for Fig 3A), indicating that the initial
without computation. The simulations indicated that the number of
cost of futile response disappeared. These concurrent fluorescence
alleles exceeding a certain frequency depended strongly (sometimes
and fitness changes agreed with the leftward hill climb on the
non-monotonically) on the overall mutation rate as well as the
blue landscape in Fig 1B (black arrow underneath D2Z0) expected
availability of individual mutations (Appendix Fig S3A). The depen-
for K-type mutations.
dence of allele numbers on simulation parameters should allow
To uncover the genetic mechanism(s) underlying these fluores-
parameter estimation once experimental allele data are available.
cence and fitness changes, we combined whole-genome and Sanger
In summary, based on mathematical and computational models,
sequencing (see the Appendix). Our analysis revealed four compet-
we hypothesized that the ancestral PF gene circuit should disappear
ing mutations inside the rtTA coding sequence that jointly
from the population fastest in conditions D2Z0 and D0Z2, followed
accounted for most of replicate population #1 already at Day 9
by D2Z2 and DiZ2, and lastly DiZ0. In addition, we conjectured that
(Fig 3C and D "12 h-1"; Appendix Table S3), and eliminated the
K, T, and G mutations should predominate in D2Z0, D2Z2, and
ancestral genotype by the end of the experiment. The same
D0Z2, respectively, whereas mixtures of T and G genotypes should
happened in the other two replicate experiments as well (Fig 3C–E
prevail in DiZ2. Mutations (K or sometimes T) should be slow to
"12 h-2,3"; Appendix Table S3). This is consistent with computa-
establish in DiZ0, causing the ancestral genotype to remain in the
tionally predicted K-type mutations eliminating rtTA toxicity, along
majority even at 20 days. To test these hypotheses, we evolved
with its transcription-activating function. We detected no mutations
three replicate PF yeast cell populations in five conditions (DiZ0,
in other parts of the genome, although we cannot rule out the possi-
D2Z0, D0Z2, DiZ2, D2Z2) corresponding to the colored squares on
bility of mutations in repeat regions or large duplications/deletions
the population fitness landscape in Fig 1B. We also evolved cells in
that are notoriously difficult to detect by whole-genome sequencing
the control condition D0Z0, where we found only one barely detect-
(Appendix Fig S4D and E). We repeated the evolution experiment
able, low-frequency synonymous substitution (Appendix Table S2).
with 24-h resuspensions and observed similar fluorescence and
We observed directly the relationship between gene expression
fitness changes, along with rtTA coding sequence mutations, except
and fitness by daily fluorescence and cell count measurements over
that they occurred faster (Fig 3C "24 h-1,2,3"; Appendix Fig S4A–C,
the course of these experiments. For various experiments and on
Appendix Table S3). Four of these mutations (three STOP codons
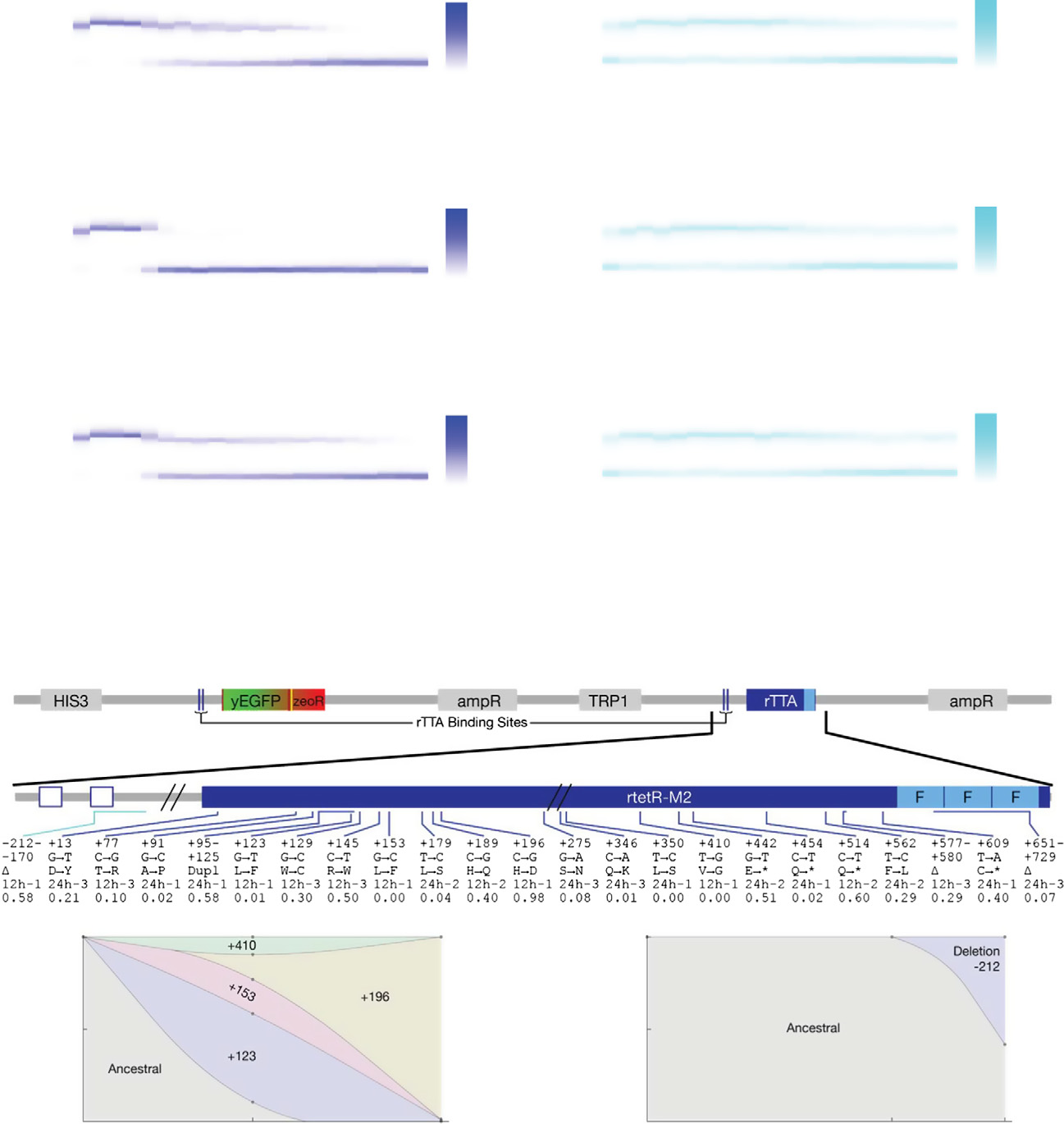
Figure 3. Evolutionary dynamics of PF cells in D2Z0 and DiZ0, corresponding to scenario (i): futile response to harmless signal.
Time-dependent changes in the fluorescence distributions (blue heatmaps), average fluorescence (blue circles), and average, mixed population fitness (bluesquares). Data were collected as PF cells evolved in condition D2Z0 (2 lg/ml doxycycline and no zeocin) in three replicate experiments. Average fluorescence and
fitness values in control condition D0Z0 are also shown as black crosses for reference. Both the fluorescence (P = 0.00019) and fitness (P = 0.003959) weresignificantly different in populations evolving in D2Z0 compared at Days 4 and 21 (t-test, see the Materials and Methods).
The same measurements as in (A), but for PF cells evolving in condition DiZ0 (0.2 lg/ml doxycycline and no zeocin, cyan heatmaps) in three replicate experiments.
The fluorescence (P = 0.0144526) was significantly different, but the fitness (P = 0.2459) was not in populations evolving in DiZ0 when compared at days 4 and 21.
Pairwise comparisons with the same days in D0Z0 showed no significant fitness differences (see the source data).
Intra-rtTA mutations observed in conditions D2Z0 (blue lines) and DiZ0 (light blue lines) mapped along the rtTA activator within the PF gene circuit sequence. Thefive lines of annotation indicate the following: (i) basepair coordinates relative to the rtTA translation start site (+1); (ii) nucleotide substitution; (iii) amino acidsubstitution; (iv) in which experiment the allele was found; and (v) allele fractions at Day 19 inferred from sequencing. If there was a deletion or duplication, thefirst two lines represent its range. *: STOP codon; D: deletion; Dupl: duplication. No extra-rtTA mutations were identified in these conditions. Clones selected forphenotyping are underlined and numbered in blue.
D, E Time-dependent allele frequencies for mutations observed in conditions D2Z0 (D), and DiZ0 (E), replicate experiment #1. The way we used sequencing data to
draw allele frequencies and the lines connecting them is explained in the Mutation time course reconstruction section of the Materials and Methods.
F, G Time-dependent allele frequencies from simulations using mutation parameter values reflecting experimental conditions.
Source data are available online for this figure.
Molecular Systems Biology
ª 2015 The Authors
Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
Gene expression & fitness, D2Z0
Gene expression & fitness, DiZ0
Day of Experiment
Day of Experiment
Day of Simulation
Day of Simulation
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
and a 78-base pair deletion) truncated and eliminated all three acti-
mutations in either rtTA or its regulatory region. Instead, we
vator domains of rtTA, further supporting the K-type loss of rtTA
detected two extra-rtTA, but intra-circuit mutations overall in six
replicate experiments (Fig 4D; Appendix Table S4), one of which
Decreasing the inducer (doxycycline) concentration from 2 to
eliminated a tetO2 operator site upstream from yEGFP::zeoR, while
0.2 lg/ml should diminish rtTA toxicity. Selection in this condition
the other was a synonymous substitution in an arginine codon
should be weaker (Fig 1B), lowering the chances of beneficial
within the zeoR coding region. Additionally, sequencing revealed
mutations establishing in DiZ0 compared to D2Z0. To test these
multiple extra-circuit mutations (Fig 4B) and linkage between the
predictions, we evolved three cultures in the DiZ0 condition
intra-circuit tetO2 deletion and some extra-circuit alleles (Fig 4B and
(Fig 3B). In agreement with computational predictions (Fig 2A and
D). This raised the possibility that intra- and extra-circuit mutations
B; Appendix Figs S2 and S3), the fraction of On cells started declin-
jointly detoxify the cells in a manner consistent with G-type muta-
ing slowly only toward the end of the experiment. This resulted in a
tions. The real number of extra-circuit mutations could be higher,
statistically significant change in fluorescence, but not in fitness.
considering the difficulty of detecting certain mutation types by
Moreover, Sanger sequencing at the end of the experiment revealed
high-throughput sequencing. In addition, some adaptation in D0Z2
a single intra-circuit deletion at 58% frequency (Fig 3C), which
could also have occurred through native stress responses or non-
eliminated one of the two tetO2 operator sites upstream of rtTA.
genetic selection of the high-expressing tail of the basal yEGFP::
This suggests a T-type mutation (since one tetO2 site remained
ZeoR distribution.
intact) targeting a regulatory region rather than protein-coding
Altogether, these data suggest that as long as cells with a
sequence. We detected no mutations elsewhere in the genome.
potentially beneficial, but inoperative module have some basal
In summary, these experimental observations confirmed the
resistance to survive, they can later activate the module and
computational predictions that a steep, monotonically decreasing
acquire drug resistance by genetic mutations (Charlebois et al,
cellular fitness landscape (Fig 1B, D2Z0, blue shading; Fig 3F and G)
2011). Apparently, this happens through mutations both inside
reproducibly selects for lower gene expression. The effect of these
and outside of the module, genetically integrating it with the
mutations is to decrease gene expression unidirectionally by either
host. This effect seems dependent on the presence of zeoR, since
eliminating or reducing the fraction of On cells. Thus, deleterious
cells lacking this gene do not survive in D0Z2 (Appendix Fig
network activation favors mutations that prevent or reduce switch-
S5C and D). An interesting question is whether drug resistance
ing into the slow-growing On state. We selected five individual
gained from these mutations involves some cost. To answer this
genotypes (underlined with blue in Fig 3C) for testing whether their
question and test whether the mutants are indeed G-type, we
gene expression and fitness are consistent with K-type mutations
selected and characterized six individual genotypes underlined
(see below the section on phenotyping).
with red in Fig 4D (see the section on phenotyping below).
Scenario (ii): gaining gene expression for an initially
Scenario (iii): optimization of gene expression under opposing
unresponsive gene circuit
evolutionary pressures
To test what happens if a stress defense module cannot induce
To test what happens when a module responds to stress non-
when needed during harmful stress, we grew cell populations in
optimally, we exposed the cells to both inducer and antibiotic. In
2 mg/ml zeocin (D0Z2). The lack of inducer in this condition
these conditions, there is a cellular fitness peak at intermediate gene
forced all cells to be in the drug-sensitive Off state. Consequently,
expression (Fig 1B, DiZ2 and D2Z2, green and magenta shading), in
the tradeoff between elevated expression and drug resistance
contrast to the monotone cellular fitness landscapes in conditions
specific to the PF gene circuit was absent in D0Z2. Early in the
with only inducer (D2Z0 and DiZ0) or only antibiotic (D0Z2). The
course of evolution, we observed a substantial, statistically signifi-
cellular fitness peak indicates opposing selection pressures from
cant drop in population fitness compared to untreated cells
zeocin toxicity and the fitness cost of rtTA expression: zeocin selects
(Fig 4A), indicating the gene circuit's inability to respond to
for increased gene expression, while rtTA toxicity selects for dimin-
stress. Yet, some cells must have had enough drug resistance to
ished rtTA function and thus decreased gene expression (Fig 1B,
survive, because the growth rates of cultures started to recover
arrows underneath DiZ2 and D2Z2). These selection pressures act
after � 4 days (Fig 4A). At the same time, yEGFP∷ZeoR expres-
on two cell subpopulations flanking a cellular fitness peak (Fig 1B,
sion increased significantly compared to control cultures main-
D2Z2). Therefore, fitness improvement in DiZ2 and D2Z2 requires
tained in D0Z0 (Fig 4A). This difference remained statistically
adaptation toward an intermediate "sweet spot" of expression. K
significant even after correction for multiple comparisons, particu-
mutations cannot achieve this since they completely disrupt rtTA
larly toward the end of the experiment. We observed similar
trends with 24-h resuspensions (Appendix Fig S5). Thus, the
In D2Z2, average fluorescence decreased while fitness increased
evolving cell population moved repeatedly upward in gene
significantly for all replicate cultures (Fig 5A), albeit by a lesser
expression and drug resistance space, toward the cellular fitness
extent and more slowly than in D2Z0 (Fig 3A), as predicted compu-
maximum in Fig 1B (black arrow underneath D0Z2). In contrast,
tationally. Sequencing has uncovered only two competing alleles
cells lacking the zeoR gene never recovered in the same level of
from one replicate culture, each affecting a distinct PF gene circuit
zeocin, while cells with higher basal yEGFP::zeoR expression
component. Sequencing samples from the other two replicate
recovered faster (Appendix Fig S5C–E).
experiments then revealed D2Z2-specific mutations that repeatedly
Next, we sought mutations explaining the observed fluorescence
occurred in the same rtTA loci: the 50 untranslated rtTA region and
and fitness changes. In sharp contrast with D2Z0, we found no
the 225th basepair of rtTA (Fig 5C; Appendix Fig S6A and B;
Molecular Systems Biology
ª 2015 The Authors

Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
Gene expression & fitness, D0Z2
Day of Experiment
Day of Experiment
Figure 4. Evolutionary dynamics of PF cells in D0Z2, corresponding to scenario (ii): lack of response when needed.
Time-dependent changes in the fluorescence distributions (red heatmaps), average fluorescence (red circles), and average, mixed population fitness (red squares) asPF cells evolve in condition D0Z2 (no doxycycline and 2 mg/ml zeocin) in three replicate experiments. Black crosses, as in Fig 3. Both the fluorescence (P = 0.014323)and fitness (P = 0.002244) were significantly different in populations evolving in D0Z2 when compared at days 4 and 21 (dependent samples t-test, see the Materialsand Methods). In addition, at many time points, the fluorescence difference from the ancestral PF was statistically significant (independent samples t-test, see theMaterials and Methods). These statistical differences persisted even after correcting for multiple comparisons. The same was true for fitness at early time points (upto Day 6).
Time-dependent allele frequencies for mutations observed in condition D0Z2, replicate #1. Top: whole-genome sequencing from a 12-h resuspension experiment.
Bottom: whole-genome sequencing combined with Sanger sequencing of clonal isolates from the same 24 h resuspension experiment, indicating linkage betweenintra- and extra-PF mutations. Among the observed mutations, INO2 is a regulator of phospholipid biosynthesis that lowers stress resistance, and YHR127W functionis unknown, but is synthetic lethal with ARP1, which mediates resistance to multiple stresses. Red bars and numbers indicate clones selected for phenotyping. Theway we used sequencing data to draw allele frequencies and the lines connecting is explained in the Mutation time course reconstruction section of the Materialsand Methods.
C Time-dependent allele frequencies from simulations using mutation parameter values reflecting experimental observations.
D Extra-rtTA, but intra-circuit mutations observed in condition D0Z2 (red lines) mapped along yEGFP::zeoR within the PF gene circuit sequence. The five lines of
annotation indicate the following: (i) basepair coordinates relative to the yEGFP::zeoR translation start site (+1); (ii) nucleotide substitution; (iii) amino acidsubstitution; (iv) which experiment the allele was found; and (v) allele fractions at Day 19 inferred from sequencing. If there was a deletion, the first two linesrepresent its range. D, deletion; Syn, synonymous. Two extra-rtTA, but intra-circuit mutations were identified in this condition. Clones selected for phenotyping areunderlined and numbered in red.
Source data are available online for this figure.
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
Appendix Table S5). Another mutation truncated rtTA by a STOP
measured gene expression levels and population fitness of clonal
codon in the last activator domain but left the two other domains
isolates from the last day of the evolution experiments.
intact, suggesting a T mutation with diminished rtTA function and
First, we studied five clonal isolates from the last day of the
toxicity, while still maintaining a zeocin-resistant, yEGFP∷ZeoR-
D2Z0 time course (underlined in blue in Fig 3C), to test whether
they carry K-type mutations. If this is true, then they should be
Next, we studied how lower but nonzero rtTA toxicity affects
uninducible and their fitness should not depend on doxycycline.
evolutionary dynamics for a peaked fitness landscape, propagating
Therefore, we quantitatively characterized the effect of doxycycline
PF cells in 0.2 lg/ml doxycycline and 2 mg/ml zeocin (Fig 1B,
on the fluorescence and fitness of these clones. Thus, we defined
DiZ2). The addition of zeocin selects against low-expressing Off
the fitness effect of doxycycline as log10[(fitness with doxycycline)/
cells, reshaping the bimodal distribution seen in DiZ0, so that the
(fitness without doxycycline)]. Likewise, we defined the effect of
fraction of On cells increases in DiZ2 (compare black histograms
doxycycline on fluorescence as log10[(fluorescence with doxy-
overlaid with cyan and green shading, DiZ0 and DiZ2 in Fig 1B).
cycline)/(fluorescence without doxycycline)]. Based on these
These high expressors thus survive in stress and can maintain the
measures, we found that all five clones isolated from the D2Z0
population until more potent drug resistance mutations arise
inducer-only condition were fitter (Fig 6A, top panel) than the PF
(Charlebois et al, 2011). Indeed, fitness decreased only slightly
ancestor and were uninducible (Fig 6A, middle panel). These
during evolution in DiZ2 (Fig 5B). After Day 7, fluorescence seemed
properties matched the characteristics of K-type mutations predicted
to decrease slowly while fitness crept up throughout the time
computationally to dominate in D2Z0 (Fig 6A). Sanger sequencing
course. These changes were not statistically significant when we
of clonal isolates from the middle and the end of the D2Z0 evolution
compared fitness and fluorescence values at Day 4 and Day 21 along
time course indicated that each K-type mutation occurred individu-
the DiZ2 time course. However, we found that the fitness in DiZ2
ally, without linkage to other mutations. Some of these clones were
was significantly lower than in D0Z0 at several time points, which
also fitter in D0Z0 compared to the ancestral strain, suggesting
remained true even after correcting for multiple comparisons.
additional adaptation to growth in minimal medium (Lenski &
DiZ2 was the only condition where mutations affecting both rtTA
Travisano, 1994; New et al, 2014) after eliminating the rtTA toxicity.
and extra-circuit loci established (Fig 5E). The intra-circuit mutation
Next, we studied clonal isolates from the last day of the D0Z2
was a tetO2 site deletion from the rtTA promoter, eliminating the
time course (Fig 4B and D) to test whether they are zeocin-resistant.
other tetO2 site compared to the deletion in DiZ0. Additionally, we
We quantitatively characterized the effect of zeocin on the fitness of
detected three extra-rtTA mutations, one of them linked to the tetO2
these clones as log10[(fitness with zeocin)/(fitness without zeocin)].
deletion. In general, these findings indicated that peaked fitness
To determine whether zeocin resistance arose from higher yEGFP::
landscapes selected for T-type mutations, while also allowing
zeoR expression, we also defined the gene expression increase in
for G-type mutations, as predicted computationally (Figs 2D
these clones as log10[(fluorescence of evolved clone in D0Z0)/(fluo-
and 5F and D). We confirmed these mutation types by testing
rescence of PF ancestor in D0Z0)]. We found that all clones isolated
whether the mutations weakened rtTA activity, without eliminating
from the zeocin-only condition (underlined in red in Fig 4B and D)
it (see below).
had higher fitness in zeocin (D0Z2) compared to ancestral PF cells(Fig 6B, top panel). The cause of zeocin resistance was higher
Phenotyping reveals fitness-improving network characteristics
yEGFP::zeoR gene expression even in the condition D0Z0, withoutzeocin (Fig 6B, middle panel and Fig 6E). These observations are
In contrast to the D0Z0 control condition where fitness and gene
consistent with G-type mutations, predicted computationally to
expression changes were statistically non-significant (Appendix Fig
dominate in D0Z2. yEGFP::zeoR gene expression in all clones shifted
S6D and E), these quantities changed significantly in other condi-
significantly upward, obeying the selection pressure (Fig 1B, black
tions tested (Figs 3, 4 and 5). These changes generally involved
arrow underneath D0Z2). Some clones had two linked mutations,
mixed populations composed of different genotypes competing with
one of which was within the PF gene circuit, while the other was
each other. To characterize individual genotypes in isolation, we
outside of it. We found no mutations for one zeocin-resistant clone
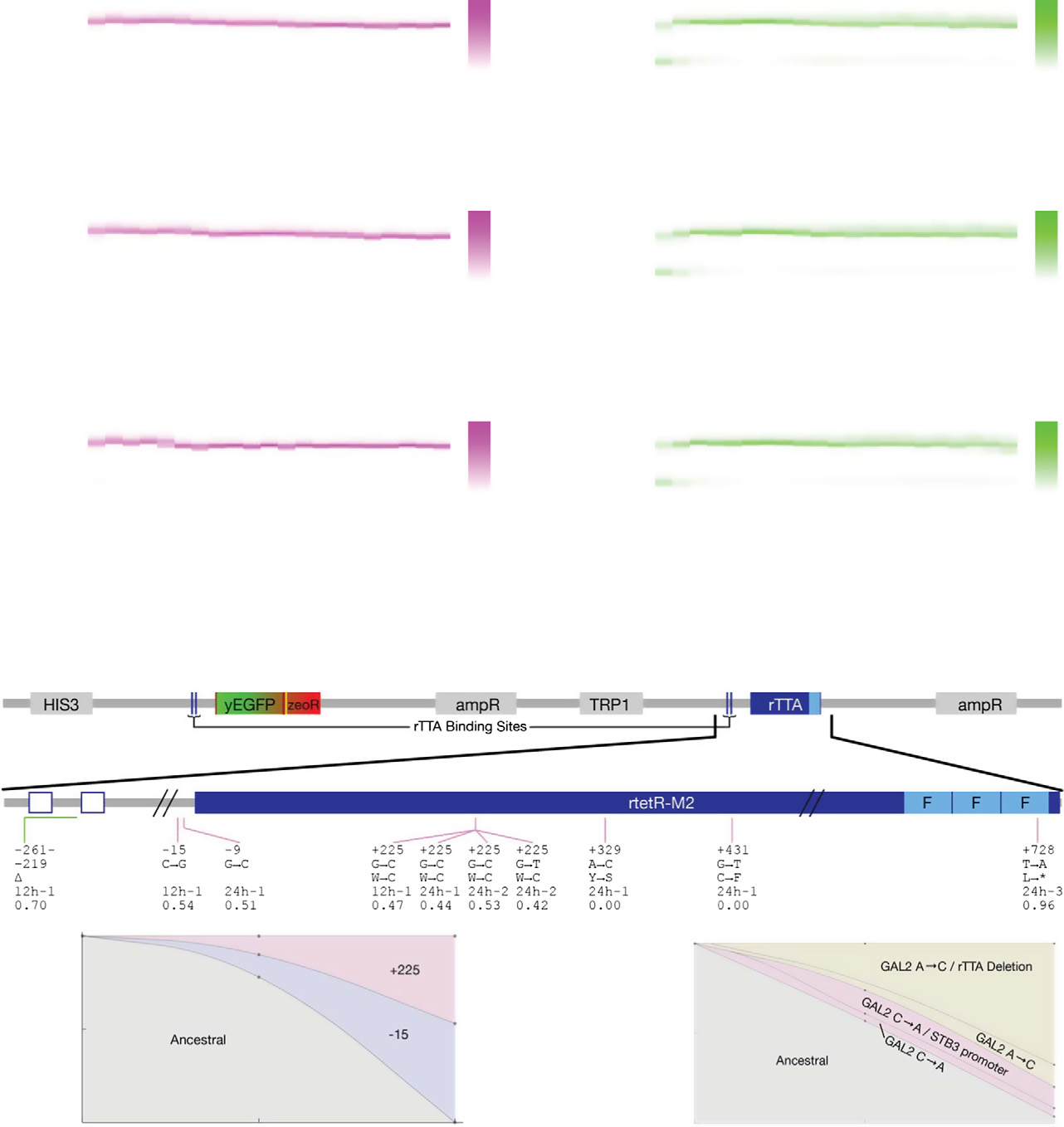
Figure 5. Evolutionary dynamics of PF cells in D2Z2 and DiZ2, corresponding to scenario (iii): suboptimal response.
Time-dependent fluorescence distributions (magenta heatmaps), average fluorescence (magenta circles), and mixed population fitness (magenta squares) as PFcells evolve in condition D2Z2 in three replicate experiments. Black crosses, same as in Fig 3. Both the fluorescence (P = 0.0003157) and fitness (P = 0.010568) weresignificantly different in populations evolving in D2Z2 when compared at days 4 and 21. Statistical test: as above.
The same measurements as in panel (A), but for PF cells evolving in condition DiZ2 in three replicate experiments. Neither fluorescence (P = 0.95), nor fitness(P = 0.087) was significantly different in populations evolving in DiZ2 when compared at days 4 and 21. Pairwise comparisons with the same days in D0Z0 showedsignificant fitness differences, many of which remained true even after correction for multiple comparisons.
Intra-circuit mutations observed in conditions D2Z2 (magenta lines) and DiZ2 (green lines) mapped along the rtTA activator within the PF gene circuit sequence.
The five lines of annotation indicate: (i) basepair coordinates relative to the rtTA translation start site (+1); (ii) nucleotide substitution; (iii) amino acid substitution;(iv) which experiment the allele was found; and (v) allele fractions at Day 19 inferred from sequencing. If there was a deletion, the first two lines represent itsrange. *, STOP codon; D, deletion. While no extra-rtTA mutations were identified in condition D2Z2, a few were found in DiZ2 (see E). Clones selected forphenotyping are underlined and numbered.
D, E Time-dependent allele frequencies for mutations observed in conditions D2Z2 (D) and DiZ2 (E), replicate #1. The way we used sequencing data to draw allele
frequencies and the lines connecting is explained in the Mutation time course reconstruction section of the Materials and Methods.
F, G Time-dependent allele frequencies from simulations using mutation parameter values reflecting experimental conditions.
Source data are available online for this figure.
Molecular Systems Biology
ª 2015 The Authors
Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
Gene expression & fitness, D2Z2
Gene expression & fitness, DiZ2
Day of Experiment
Day of Experiment
Day of Simulation
Day of Simulation
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
in any locus tested by Sanger sequencing, suggesting extra-circuit
placed in D2Z2 were not. Taken together, these observations suggest
mutation(s) undetectable by either whole-genome or targeted
that gene circuit bistability may have trapped mutant cells in the On
Sanger sequencing. These results indicate that adaptation in D0Z2
state during evolution if the mutation arose in cells that were On.
recurrently involves mutations causing PF gene expression increase.
Finally, we studied the single clone isolated from the DiZ2 exper-
This is surprising considering that mutations could have just upreg-
iments (tetO2 deletion; underlined in green, Fig 5C). These cells
ulated native stress resistance pathways without involving the PF
required only slightly higher doxycycline levels for induction than
gene circuit.
the PF ancestor. Yet, once induced, they rose to higher mean expres-
Considering the original tradeoff between the cost of gene expres-
sion level than the ancestor (Fig 6D; Appendix Fig S7A). Moreover,
sion and benefit of drug resistance in the PF gene circuit, we asked
the two peaks in the bimodal gene expression histograms
whether a similar tradeoff may apply to drug-resistant genotypes
approached each other for this clone (Fig 6F; Appendix Fig S7A),
evolved in D0Z2. Interestingly, adaptation by elevated basal yEGFP::
both shifting toward the cellular fitness peak in Fig 1B, DiZ2 as
zeoR expression tended to cause a fitness cost in D0Z0, when zeocin
dictated by selection. This is a unique example of noisy gene expres-
was absent (Fig 6B, bottom panel). The sources of these new fitness
sion evolving under opposing selection pressures (Fig 1B, black
costs are unclear, but they are not rtTA-related because doxycycline
arrows underneath DiZ2). Essentially, although evolution altered
was absent. This suggests that a novel tradeoff appeared between
the gene expression, its distribution still remained bimodal, with a
evolved stress resistance and growth in the absence of stress
similar mean. While this mutation apparently alters rtTA function, it
(Fig 6G). This new tradeoff is reminiscent of the original tradeoff in
is different from the T-type mutations assumed in computational
the ancestral PF gene circuit, where higher expression was also
models (which did not account for shifting of peaks). This unique
costly, but protective in the presence of antibiotic.
type of adaptation has no equivalent in phenotypically homo-
We similarly characterized the effects of doxycycline and zeocin
geneous populations with unimodal gene expression distributions.
on the fitness and gene expression of two clones isolated from D2Z2
To measure the phenotypic effects of the observed mutations in
experiments (underlined in magenta, Fig 5C). We found that both
isolation from potential changes in the genetic background, we recon-
clones isolated from D2Z2 had reduced inducer sensitivities
structed the mutations rtTA+225G?C (D2Z2 clone #1) and rtTA�9G?C
(Fig 6C), requiring higher doxycycline than the PF ancestor to reach
(D2Z2 clone #2) in the ancestral PF background (Appendix Fig S7C
a given gene expression level (Fig 6C, middle panel). Generally, the
and D). The rtTA+225G?C mutation was slightly inducible in the
gene expression distributions of these clones were enriched in Off
ancestral background, with a small high-expressing subpopulation at
cells (Appendix Fig S7C and D). These changes were associated with
2 lg/ml doxycycline. Moreover, we could reinduce this clone to
lower doxycycline toxicity (Fig 6C, top panel, blue bars), while the
nearly full expression using excessive doxycycline concentrations
cells still maintained drug resistance in doxycycline (Fig 6C, bottom
(6 lg/ml) in the presence of zeocin, suggesting that the reconstructed
panel, magenta bars). These characteristics were consistent with
mutation rtTA+225G?C lowered the dynamic range and sensitivity
T-type mutations, as predicted computationally to dominate for
similar to the clonal isolate. Interestingly, however, the reconstructed
peaked cellular fitness landscapes. Interestingly, in addition to the
rtTA�9G?C mutation failed to induce even with excessive doxycyline
increase in Off cells, the On state moved to lower expression, toward
concentrations, suggesting linkage and potential epistasis with some
the cellular fitness peak in the gene expression space (Appendix Fig
undetectable genetic extra-circuit mutation(s).
S7C and D). We could still fully induce these clones by applying
Overall, phenotyping validated the prevalence of K, T, G muta-
excessive (6 lg/ml) doxycycline levels with zeocin (Appendix Fig
tion types in different environments, as predicted computationally.
S7C and D). Interestingly, all cells were in the On state (fully
Our observations also underscore the potential importance of noise-
induced) throughout the 20 days of evolution in D2Z2, but clones
reshaping T-type mutations in artificial and natural evolution.
Figure 6. Gene expression and fitness characteristics of clonal isolates from various evolved populations.
Phenotype of clones evolved in inducer doxycycline alone (D2Z0, "futile response"). The first bar ("Anc.") corresponds to the ancestral PF cells, and the other barscorrespond to clonal isolates from the last time point of the D2Z0 experiment. Top panel: log
10-ratio of fitness with doxycycline (D2Z0) relative to no doxycycline
(D0Z0). Middle panel: log10-ratio of average fluorescence intensity with doxycycline (D2Z0) relative to no doxycycline (D0Z0). Bottom panel: log10-ratio of averagepopulation fitness of each evolved clone relative to the ancestor in no doxycycline (D0Z0). Error bars represent standard deviations around the mean. Stars denotesignificance at P < 0.05 (two-sided t-test).
Phenotype of clones evolved in antibiotic zeocin alone (D0Z2, "lack of response when needed"). The first bar ("Anc.") corresponds to the ancestral PF cells, and theother bars correspond to mutants. Top panel: log10-ratio of fitness with zeocin (D0Z2) relative to no zeocin (D0Z0). Middle panel: log10-ratio of average fluorescenceintensity of each evolved clone relative to the ancestor in no zeocin (D0Z0). Bottom panel: log10-ratio of average population fitness of each evolved clone relative tothe ancestor in no zeocin (D0Z0). Error bars and stars as in (A).
C Phenotypes of two clones evolved in doxycycline and antibiotic zeocin (D2Z2, "suboptimal response"). The bars marked "A." correspond to the ancestral PF cells, and
the other bars correspond to mutants. Top panel: log10-ratio of fitness with doxycycline (D2Zy) relative to no doxycycline (D0Zy) either with or without zeocin (y = 0or y = 2). Middle panel: log10-ratio of average fluorescence intensity with doxycycline (D2Zy) relative to no doxycycline (D0Zy). Bottom panel: log10-ratio of fitnesswith zeocin (DxZ2) relative to no zeocin (DxZ0), either with or without doxycycline (x = 0 or x = 2). Error bars and stars as in (A).
D Phenotype of the single clone isolated from intermediate doxycycline and antibiotic zeocin (DiZ2, "suboptimal response"). The bars marked "A." correspond to the
ancestral PF cells, and the other bars correspond to the mutant clone. Top panel: log10-ratio of fitness with doxycycline (D2Zy) relative to no doxycycline (D0Zy) eitherwith or without zeocin (y = 0 or y = 2). Middle panel: log10-ratio of average fluorescence intensity with doxycycline (D2Zy) relative to no doxycycline (D0Zy). Bottompanel: log10-ratio of fitness with zeocin (DxZ2) relative to no zeocin (DxZ0), either with or without doxycycline (x = 0 or x = 2). Error bars and stars as in (A).
Gene expression histograms measured in D0Z0 for Clones #4 and #7 (evolved in D0Z2) compared to the PF ancestor (shaded histogram).
Gene expression histograms measured in DiZ2 for Clone #1 (evolved in DiZ2) compared to the PF ancestor (shaded histogram).
G Tradeoff between yEGFP::zeoR expression and zeocin resistance for clones evolved in D0Z2 (red) and DiZ2 (green).
Molecular Systems Biology
ª 2015 The Authors
Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
Clones evolved in D2Z0
Clones evolved in D0Z2
Clones 1, 2 evolved in D2Z2
Clone 1 evolved in DiZ2
Bar colors indicate conditions where clones were tested:
log (fitness without Zeocin)
Fluorescence in D0Z0 (arb.units)
Fluorescence in DiZ2 (arb.units)
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
Additional insights into PF evolutionary dynamics
mutations. Likewise, the pre-existing mutations did not substan-tially alter the ancestral genotype's half-life in any condition
Experimental evolution and phenotyping validated the major muta-
(Appendix Fig S8).
tion types predicted computationally for each condition. Therefore,we asked whether the computational framework could provide anyadditional insights into evolutionary forces and mechanisms based
on the experimental data.
First, we tried to estimate the rate l of potentially beneficial
Stress response networks play key roles in the emergence of drug
mutations using its predicted effect on various allele numbers in
resistance, from pathogenic microbes to cancer. Typically, stress
several conditions. Interestingly, we could not capture the experi-
response incorporates a tradeoff: cells that activate it grow slower in
mental number of alleles and the half-life of ancestral genotype
the absence of stress. Therefore, optimality of these networks
when we applied the same mutation rate in all conditions. Instead,
depends on maintaining the balance between environmental stress
comparing the results of computational simulations (Fig 4C;
and internal response. Yet, it is unknown how quickly, how repro-
Appendix Fig S2D and F) with experimental data suggested slightly
ducibly, and through what types of mutations stress response
higher mutation rate in zeocin than without it (Fig 2). Specifically,
networks evolve to balance the costs and benefits of their response
the rate of potentially beneficial mutations that matched the data
to external stress. What aspects of network evolution are predictable
best was l�Z = 10�6.2 with zeocin compared to l+Z = 10�5.4/
a priori and what is required for making predictions is unclear. To
genome/generation without zeocin. This increase is reasonable
address these questions, we studied evolving yeast cells endowed
because zeocin is a DNA-damaging agent that may elevate mutation
with a synthetic stress response gene circuit that allowed for sepa-
rates. These beneficial mutation rates are comparable with a recent
rate control of the stress and the response by adjusting antibiotic
estimate in yeast of 10�6/genome/generation (Levy et al, 2015).
and inducer concentrations, respectively.
Second, we asked whether we could extract any information
Using quantitative knowledge of the PF gene circuit, we devel-
about the mutation probabilities P(K), P(T), and P(G). We compared
oped two computational models to predict specific aspects of evolu-
simulation results with experimental data in D2Z0 and D2Z2
tionary dynamics in six different environmental conditions. The
conditions where K- and T-type mutations should be prevalent,
predicted aspects included the speed at which the ancestral geno-
respectively. Selection for various mutation types is environment-
type disappears from the population, as well as the types and
dependent, implying that the number and type of established
numbers of mutant alleles that establish in each environmental
mutations must depend on the K/T bias in mutations entering the
condition. We validated these predictions by experimental evolu-
population. For example, while mainly T-type mutations can
tion. The agreement between our predictions and experimental find-
establish and the K-type is deleterious in D2Z2 (since it forces cells
ings suggests that cellular and population fitness landscapes can be
into the drug-sensitive Off state), the opposite is true in D2Z0.
useful to predict short-term evolution. Critically, our predictive
Comparing experimentally observed allele numbers with simulation
models were based on quantitative knowledge of the fitness and
results indicated that incoming T mutations should be in the
gene expression properties, as well as the genetic structure (design)
minority (gray bars, Fig 2B and C) compared to the 10 times more
of the PF gene circuit. Without such knowledge, it would have been
available K-type mutations. This suggests that only a few, specific
impossible to predict what type of mutations arise and how fast.
rtTA loci can harbor T-type mutations, explaining recurrence of
Once this knowledge is acquired, however, cellular and population
certain mutations in D2Z2. These recurrent mutations must have the
fitness landscapes (Fig 1B) can be constructed, which are informa-
rare capability of tweaking protein function and toxicity while still
tive for predicting evolutionary outcomes.
maintaining drug resistance, as predicted computationally and vali-
We found a connection between the rates at which various
dated experimentally (Fig 6).
potentially beneficial mutations entered the populations and the
Finally, we asked how mutations that arose prior to setting the
computationally predicted features of evolutionary dynamics, espe-
environmental conditions may have contributed to the outcome of
cially the number of mutant alleles (Appendix Fig S3). This allowed
evolution experiments. This was important because the PF cells
a rough estimation of the relative probabilities of two mutation
grew for 24 h in D0Z0 before initiating our evolution experiments.
types to occur spontaneously. We found that mutations eliminating
To address this question, we used a variant of the simulation
protein function were much more common than mutations fine-
framework that allowed neutral mutations to accumulate for 24 h
tuning protein function (at least for rtTA in these experiments). The
of growth at the mutation rate l�Z = 10�6.2/genome/generation.
availability of various beneficial mutation types depends on DNA
Afterward, we changed the simulated condition to DiZ0, D2Z0,
sequence and is rarely known a priori. We suggest nonetheless that
DiZ2, D2Z2, or D0Z2 using values of the free parameters
the availability of mutation types could be estimated by comparing
estimated from experimental data. We then computed the contri-
computational predictions with actual observations in similar labo-
bution of these "preexisting" mutations to the final allele frequen-
ratory evolution experiments.
cies (Appendix Fig S8). The results indicated that preexisting
A unifying theme for all environmental conditions was the trade-
mutations do not comprise a large fraction of mutant alleles in
off between stress resistance and stress-free growth: genotypes that
conditions DiZ0, D2Z2, and DiZ2. On the other hand, in steep
resisted zeocin tended to grow slower in its absence. Such tradeoffs
monotonic cellular fitness landscapes (D2Z0 and D0Z2), preexist-
were inherent by design to the ancestral PF synthetic gene circuit
ing alleles could comprise approximately 35% of mutant alleles in
(Fig 1B). However, in D0Z2, yeast adapted using extra-PF mutations
D2Z0 and 50% in D0Z2. Nevertheless, the same mutation types
that were not subject to the original tradeoff. Most surprisingly,
dominated in specific conditions with or without pre-existing
these extra-circuit changes were subject to a different tradeoff,
Molecular Systems Biology
ª 2015 The Authors
Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
which resembled the original one in the PF gene circuit (Fig 6G).
(SD) medium with 2% weight of sugar (glucose or galactose) and
Essentially, there was a cost for higher yEGFP::zeoR expression,
the appropriate supplements (-his, -trp) to maintain auxotrophic
even if caused by extra-circuit mutations. Thus, without the built-in
selection (reagents from Sigma, St. Louis, MO).
tradeoff within the PF gene circuit, another tradeoff appears throughmutations outside of the PF gene circuit. This suggests a fundamen-
Experimental evolution
tal conflict between two different tasks (resistance to stress and fastgrowth in stress-free conditions), typically resolved by Pareto opti-
In preparation for the experiments, the PF ancestor strain was
mization (Shoval et al, 2012). Such "multi-layered" tradeoffs (when
streaked on SD 2% glucose plates. Plates were incubated at 30°C for
multiple ways of coping with stress exist, but each has its own type
2 days. Well-isolated single colonies were picked into 1 ml SD-
of tradeoff) may occur frequently in many natural systems, includ-
his-trp 2% galactose liquid medium and incubated overnight at 30°C
ing more complex genetic circuits in other organisms.
with orbital shaking at 250 rpm and resuspended regularly (every
The ultimate success of synthetic biology will depend on the
12 h or every 24 h). Fluorescence and cell density measurements
long-term practical applicability of synthetic constructs. Despite
were taken daily or every 12 h. Samples were saved daily and
the growing number of synthetic constructs, their evolutionary
stored in 80% glycerol at �80°C for further studies. Further details
stability only recently began to be investigated in Escherichia coli
are described in the Appendix.
(Yokobayashi et al, 2002; Sleight et al, 2010; Wu et al, 2014). As faras we know, this question has not been addressed in eukaryotes.
Fitness landscape mapping and parameter estimation
Our work fills this gap and generates insights for building evolution-arily robust eukaryotic gene circuits. The PF gene circuit is based on
Ancestral PF cells were prepared as described above. Cultures
the rtTA activator, which is widely utilized in eukaryotic synthetic
were then resuspended into the following treatments: zeocin only
biology. An important insight that we gained was that eukaryotic
(0.5, 1.0, 1.5 and 2.0 mg/ml), doxycycline only (0.2, 0.5, 1 and
activators like rtTA are not ideal if gene circuit stability is a concern.
2 lg/ml), and both doxycycline (0.2, 0.5, 1 and 2 lg/ml) and zeocin
There is evidence that eukaryotic activators are generally toxic
(2 mg/ml). Cell density and fluorescence were measured every 6 h
(Baron et al, 1997), which seems to be true for some prokaryotic
components as well (Tan et al, 2009). To address this problem,
Population and cellular growth rates were estimated using mathe-
some groups have tried to identify eukaryotic activators with
matical models described previously (Nevozhay et al, 2012) and as
reduced toxicity (Baron et al, 1997; Khalil et al, 2012). Still, we
described in the Appendix. Briefly, we used fitness functions to
would recommend avoiding long-term use of common eukaryotic
model the effects of conditions and gene expression on growth. One
activators (utilizing VP16, VP64, or GAL activator domains, includ-
depends on zeocin and yEGFP::ZeoR protein concentration:
ing in dCas9-, TALE-, or zinc finger-based synthetic regulators) until
vþziðF;ZÞ where Zi is inferred from the steady-state solution of a
their genetic stability has been carefully tested in long-term evolu-
tion experiments. Our experiments could be considered as testingrtTA activator stability in various environments. The experiments
_Zi ¼ /Z � hzZi � sRZi
revealed the evolutionary instability of rtTA, but also led to the
discovery of mutant activators and gene circuit designs with loweractivator toxicity. These could become novel parts and designs mini-
with Z, B, and R representing external zeocin, and bound and
mizing activator toxicity when eukaryotic activators are needed, as
unbound yEGFP::ZeoR protein concentrations (F = B + R). The
in memory circuits (Ajo-Franklin et al, 2007; Burrill et al, 2012).
other depends on doxycycline and yEGFP::ZeoR protein concentra-
To conclude, this work highlights the unique ability of synthetic
tion, assumed to be equal with rtTA protein concentration:
biological constructs to provide improved, quantitative understand-
with C representing doxycycline concentration. The
ing and predictability to fundamental biological processes such as
total growth rate is then c = c1c2.
evolution and development. Similar studies will be essential to
In each condition, the rate of switching from low to high expres-
assess and improve the evolutionary stability of synthetic gene
sion and vice versa (cellular memory) was inferred from experimen-
circuits, enabling their industrial and clinical application. Therefore,
synthetic biology is about to reverse the information flow toward
(Nevozhay et al, 2012; Appendix Fig S3E).
other fields of biology, the source of original inspiration for parts
Resulting parameter estimates are presented in Appendix
and concepts for the first synthetic genetic constructs.
Statistical analysis of gene expression and fitness data
Materials and Methods
Fluorescence and fitness values were compared using t-tests in our
Strains and media
study. We used an "independent samples" version of the t-test tocompare different conditions (for example, D0Z0 and D0Z2). On the
We used the haploid Saccharomyces cerevisiae strain YPH500
other hand, we used a dependent (paired) samples version of the
(a, ura3-52, lys2-801, ade2-101, trp1D63, his3D200, leu2D1; Stratagene,
t-test to compare different time points within one environmental
La Jolla, CA) with the PF synthetic gene circuit stably integrated into
condition. We applied Bonferroni correction for multiple compar-
chromosome XV near the HIS3 locus as described previously
isons whenever applicable. All tests were performed in STATISTICA
(Nevozhay et al, 2012). Cultures were grown in synthetic dropout
9.1 (StatSoft Inc., Tulsa, OK).
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
Gene expression and fitness characterization of clonal
In many conditions, we observed multiple mutations in the same
isolates (phenotyping)
sample. To obtain information on linkage, we performed Sangersequencing on clonal isolates, at mutation loci determined by
Fitness of clones isolated from evolved populations was estimated
whole-genome Illumina sequencing. When linked mutations were
using an Infinite M200 Pro plate reader (Tecan) for OD600 measure-
called, we averaged the whole-genome frequency estimates of the
ments (600 � 9 nm, number of reads = 25) of orbitally shaken
two mutants to approximate the linked allele frequency. We then
(280.8 rpm with amplitude 2 mm) 250 ll cultures in 96-well plates
averaged that value with frequency estimates from Sanger sequenc-
at 30 � 0.5°C. Cultures were rediluted into fresh media of identical
ing. This method permitted inference of linked-mutant frequencies
composition every 12 h. Fluorescence was measured every 24 h by
at time points that used both sequencing methods. For example,
flow cytometry.
Appendix Fig S4B shows just the whole-genome inferred allelefrequencies for all whole-genome-sequenced time points with no
Mathematical and computational models
linkage data (thus erroneously indicating lack of linkage for alldetected alleles). To illustrate the likely course of allele dynamics at
We have developed two different types of predictive models. The
times between the measured points, we used second-order spline
first model was a set of ordinary differential equations (ODEs) with
interpolation (gray lines).
the number of ancestral cells, and mutants lumped into K, T, G cate-gories as variables, assuming constant population size. A detailed
Expanded View for this article is available online:
description of the model is in the Appendix.
The second model was an evolutionary simulation framework
explicitly accounting for each individual mutation over the time
course written in Python 3. In the framework, we used a linear
This research was supported by the NIH Director's New Innovator Award
system of ordinary differential equations (ODEs) to describe ances-
Program (1DP2 OD006481-01), by NSF/IOS 1021675 and the Laufer Center
tral and mutant cells, with experimentally inferred growth rates
for Physical & Quantitative Biology to GB and an Alfred P. Sloan Research
(gL, gH) and switching rates (r, f). The simulation framework
Fellowship to AVM. DN acknowledges support from Program # 1326 of
includes population growth, zeocin internalization dynamics, entry
the Ministry of Education and Science, Russian Federation, and CG
of mutation types K, T, or G into the population and simulated
acknowledges support from the Division of Academic Affairs at the MD
12-h resuspensions (Appendix Fig S1B and C). We simulated all of
Anderson Cancer. Sequencing was performed at MD Anderson's DNA
the experimental conditions with appropriate growth and switching
Analysis core facility (funded by NCI CA16672). We would like to thank
parameters. To test the effect of preexisting mutations, we
the organizers and participants of the NSF-supported (Grant #1066293)
simulated a 24-h period without selection before changing the
Aspen Center for Physics workshop "Evolutionary Dynamics and Informa-
parameters to those appropriate for each condition. The simulation
tion Hierarchies in Biological Systems" (2012) and the NSF-supported
framework is described in greater detail in the Appendix along with
(Grant #PHY11-25915) "Cooperation and the Evolution of Multicellularity"
its Python script.
workshop (2013, Kavli Institute for Theoretical Physics) for discussions. MM
The rates of switching, growth, and zeocin internalization were all
would like to thank Bill Flynn and Ariella Sasson for assistance with
determined experimentally prior to the simulations (Appendix
sequencing data analysis. We thank J. Xavier, M. Rosner, D. Charlebois, M.
Table S1). Thus, the three free parameters in each condition were
Szenk, T. Székely, S. Levy and J. J. Collins for comments and discussions.
beneficial mutation rate (l), and the relative probabilities of a mutation
We also thank two anonymous reviewers for their highly insightful and
being of type K, or T. These parameters were systematically scanned
in both models to determine the robustness of our predictions.
Author contributions
Mutation time course reconstruction
CG, JCJR, MM, RMA, DN, AVM, and GB designed research; CG and DN performed
experiments; CG, JCJR, MM, RMA, DN, and GB analyzed the data; CG, JCJR, MM,
We reconstructed time courses of mutation frequencies for experi-
RMA, DN, AVM, and GB developed computational models; and CG, JCJR, MM,
mental evolution replicates D2Z0-12 h-1 (Fig 3D), DiZ0-12 h-1
RMA, DN, AVM, and GB wrote the paper.
(Fig 3E), D0Z2-12 h-1 (Fig 4B), D2Z2-12 h-1 (Fig 5D), DiZ2-12 h-1(Fig 5E), D2Z0-24 h-1 (Appendix Fig S4B), D0Z2-24 h-1 (Fig 4B,
Conflict of interest
Appendix Fig S5B), and D2Z2-24 h-1 (Appendix Fig S6B). In each
The authors declare that they have no conflict of interest.
case, we had allele frequencies inferred from either Sangersequencing alone, whole-genome Illumina sequencing alone, orboth. For time points with allele frequency data from both meth-
ods, we plotted the mean of frequencies between the methods. Ifonly one sequencing method was applied for a given time point,
Ajo-Franklin CM, Drubin DA, Eskin JA, Gee EP, Landgraf D, Phillips I, Silver PA
we used the corresponding inferred allele frequency. The number
(2007) Rational design of memory in eukaryotic cells. Genes Dev 21:
of sequenced time points varied between 2 and 6 depending on
the condition (excluding t = 0 h; gray points). Once the ancestral
Andersson DI, Levin BR (1999) The biological cost of antibiotic resistance. Curr
genotype reached 0%, we kept it at 0% (even if in rare cases
Opin Microbiol 2: 489 – 493
mutant alleles could not account for 100% of the population
Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial
persistence as a phenotypic switch. Science 305: 1622 – 1625
Molecular Systems Biology
ª 2015 The Authors
Published online: August 31, 2015
Caleb González et al
Regulatory network evolution
Molecular Systems Biology
Balázsi G, van Oudenaarden A, Collins JJ (2011) Cellular decision making and
Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, Sherlock G (2015)
biological noise: from microbes to mammals. Cell 144: 910 – 925
Quantitative evolutionary dynamics using high-resolution lineage tracking.
Baron U, Gossen M, Bujard H (1997) Tetracycline-controlled transcription in
Nature 519: 181 – 186
eukaryotes: novel transactivators with graded transactivation potential.
Lobkovsky AE, Koonin EV (2012) Replaying the tape of life: quantification of
Nucleic Acids Res 25: 2723 – 2729
the predictability of evolution. Front Genet 3: 246
Beaumont HJ, Gallie J, Kost C, Ferguson GC, Rainey PB (2009) Experimental
Lopez-Maury L, Marguerat S, Bahler J (2008) Tuning gene expression to
evolution of bet hedging. Nature 462: 90 – 93
changing environments: from rapid responses to evolutionary adaptation.
Becskei A, Seraphin B, Serrano L (2001) Positive feedback in eukaryotic gene
Nat Rev Genet 9: 583 – 593
networks: cell differentiation by graded to binary response conversion.
Mason J, Linsay PS, Collins JJ, Glass L (2004) Evolving complex dynamics in
EMBO J 20: 2528 – 2535
electronic models of genetic networks. Chaos 14: 707 – 715
Burger RM (1998) Cleavage of nucleic acids by bleomycin. Chem Rev 98:
Maynard ND, Birch EW, Sanghvi JC, Chen L, Gutschow MV, Covert MW (2010)
A forward-genetic screen and dynamic analysis of lambda phage host-
Burrill DR, Inniss MC, Boyle PM, Silver PA (2012) Synthetic memory circuits
dependencies reveals an extensive interaction network and a new anti-
for tracking human cell fate. Genes Dev 26: 1486 – 1497
viral strategy. PLoS Genet 6: e1001017
Charlebois DA, Abdennur N, Kaern M (2011) Gene expression noise facilitates
Moon TS, Lou C, Tamsir A, Stanton BC, Voigt CA (2012) Genetic programs
adaptation and drug resistance independently of mutation. Phys Rev Lett
constructed from layered logic gates in single cells. Nature 491:
Dekel E, Alon U (2005) Optimality and evolutionary tuning of the expression
Munsky B, Neuert G, van Oudenaarden A (2012) Using gene expression noise
level of a protein. Nature 436: 588 – 592
to understand gene regulation. Science 336: 183 – 187
van Ditmarsch D, Boyle KE, Sakhtah H, Oyler JE, Nadell CD, Deziel E,
Nevozhay D, Adams RM, Van Itallie E, Bennett MR, Balázsi G (2012) Mapping
Dietrich LE, Xavier JB (2013) Convergent evolution of hyperswarming
the environmental fitness landscape of a synthetic gene circuit. PLoS
leads to impaired biofilm formation in pathogenic bacteria. Cell Rep 4:
Comput Biol 8: e1002480
Nevozhay D, Zal T, Balázsi G (2013) Transferring a synthetic gene circuit from
Elowitz MB, Leibler S (2000) A synthetic oscillatory network of transcriptional
yeast to mammalian cells. Nat Commun 4: 1451
regulators. Nature 403: 335 – 338
New AM, Cerulus B, Govers SK, Perez-Samper G, Zhu B, Boogmans S,
Gardner TS, Cantor CR, Collins JJ (2000) Construction of a genetic toggle
Xavier JB, Verstrepen KJ (2014) Different levels of catabolite repression
switch in Escherichia coli. Nature 403: 339 – 342
optimize growth in stable and variable environments. PLoS Biol 12:
Gari E, Piedrafita L, Aldea M, Herrero E (1997) A set of vectors with a
tetracycline-regulatable promoter system for modulated gene expression
Poelwijk FJ, de Vos MGJ, Tans SJ (2011) Tradeoffs and optimality in the
in Saccharomyces cerevisiae. Yeast 13: 837 – 848
evolution of gene regulation. Cell 146: 462 – 470
Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G,
Purnick PE, Weiss R (2009) The second wave of synthetic biology: from
Botstein D, Brown PO (2000) Genomic expression programs in the
modules to systems. Nat Rev Mol Cell Biol 10: 410 – 422
response of yeast cells to environmental changes. Mol Biol Cell 11:
Quan S, Ray JCJ, Kwota Z, Duong T, Balázsi G, Cooper TF, Monds RD (2012)
Adaptive evolution of the lactose utilization network in experimentally
Gatignol A, Durand H, Tiraby G (1988) Bleomycin resistance conferred by a
evolved populations of Escherichia coli. PLoS Genet 8: e1002444
drug-binding protein. FEBS Lett 230: 171 – 175
Sanchez A, Golding I (2013) Genetic determinants and cellular constraints in
Hillen W, Berens C (1994) Mechanisms underlying expression of Tn10
noisy gene expression. Science 342: 1188 – 1193
encoded tetracycline resistance. Annu Rev Microbiol 48: 345 – 369
Shoval O, Sheftel H, Shinar G, Hart Y, Ramote O, Mayo A, Dekel E, Kavanagh
Hsieh AC, Moasser MM (2007) Targeting HER proteins in cancer therapy and
K, Alon U (2012) Evolutionary trade-offs, pareto optimality, and the
the role of the non-target HER3. Br J Cancer 97: 453 – 457
geometry of phenotype space. Science 336: 1157 – 1160
Hsu C, Scherrer S, Buetti-Dinh A, Ratna P, Pizzolato J, Jaquet V, Becskei A
Sleight SC, Bartley BA, Lieviant JA, Sauro HM (2010) Designing and
(2012) Stochastic signalling rewires the interaction map of a multiple
engineering evolutionary robust genetic circuits. J Biol Eng 4: 12
feedback network during yeast evolution. Nat Commun 3: 682
Stricker J, Cookson S, Bennett MR, Mather WH, Tsimring LS, Hasty J (2008)
Kashtan N, Alon U (2005) Spontaneous evolution of modularity and network
A fast, robust and tunable synthetic gene oscillator. Nature 456:
motifs. Proc Natl Acad Sci USA 102: 13773 – 13778
Kauffman S (1993) Origins of Order: Self-Organization and Selection in
Sumner ER, Avery SV (2002) Phenotypic heterogeneity: differential stress
Evolution. New York, NY: Oxford University Press
resistance among individual cells of the yeast Saccharomyces cerevisiae.
Khalil AS, Lu TK, Bashor CJ, Ramirez CL, Pyenson NC, Joung JK, Collins JJ
Microbiology 148: 345 – 351
(2012) A synthetic biology framework for programming eukaryotic
Tan C, Marguet P, You L (2009) Emergent bistability by a growth-modulating
transcription functions. Cell 150: 647 – 658
positive feedback circuit. Nat Chem Biol 5: 842 – 848
Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai
Tanouchi Y, Pai A, Buchler NE, You L (2012a) Programming stress-induced
MM (2013) Pervasive genetic hitchhiking and clonal interference in forty
altruistic death in engineered bacteria. Mol Syst Biol 8: 626
evolving yeast populations. Nature 500: 571 – 574
Tanouchi Y, Smith RP, You L (2012b) Engineering microbial systems to
Lenski RE, Travisano M (1994) Dynamics of adaptation and diversification: a
explore ecological and evolutionary dynamics. Curr Opin Biotechnol 23:
10,000-generation experiment with bacterial populations. Proc Natl Acad
Sci USA 91: 6808 – 6814
Tenaillon O, Rodriguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long
Levy SF, Ziv N, Siegal ML (2012) Bet hedging in yeast by heterogeneous,
AD, Gaut BS (2012) The molecular diversity of adaptive convergence.
age-correlated expression of a stress protectant. PLoS Biol 10: e1001325
Science 335: 457 – 461
ª 2015 The Authors
Molecular Systems Biology
Published online: August 31, 2015
Molecular Systems Biology
Regulatory network evolution
Caleb González et al
Thattai M, van Oudenaarden A (2004) Stochastic gene expression in
effect on global gene expression in Saccharomyces cerevisiae. Yeast 22:
fluctuating environments. Genetics 167: 523 – 530
Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R (2012) Evolutionary
Wu F, Menn DJ, Wang X (2014) Quorum-sensing crosstalk-driven synthetic
paths to antibiotic resistance under dynamically sustained drug selection.
circuits: from unimodality to trimodality. Chem Biol 21: 1629 – 1638
Nat Genet 44: 101 – 105
Yamaguchi Y, Park JH, Inouye M (2011) Toxin-antitoxin systems in bacteria
Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W (2000)
and archaea. Annu Rev Genet 45: 61 – 79
Exploring the sequence space for tetracycline-dependent transcriptional
Yokobayashi Y, Weiss R, Arnold FH (2002) Directed evolution of a genetic
activators: novel mutations yield expanded range and sensitivity. Proc Natl
circuit. Proc Natl Acad Sci USA 99: 16587 – 16591
Acad Sci USA 97: 7963 – 7968
Wang YH, Arenas CD, Stoebel DM, Cooper TF (2013) Genetic background
License: This is an open access article under the
affects epistatic interactions between two beneficial mutations. Biol Lett 9:
terms of the Creative Commons Attribution 4.0
License, which permits use, distribution and reproduc-
Wishart JA, Hayes A, Wardleworth L, Zhang NS, Oliver SG (2005) Doxycycline,
tion in any medium, provided the original work is
the drug used to control the tet-regulatable promoter system, has no
properly cited.
Molecular Systems Biology
ª 2015 The Authors
Source: http://www.buschboerries-lab.de/wp-content/uploads/2015/10/827.full_.pdf
Centre for Christian Ethics 120 Herring Rd, Eastwood NSW 2122 OCCASIONAL PAPER 7 - REVISED Should RU486 be available in Australia? "There is no quick fix for pregnancy, no magic pill." – father of 18-year-old Californian woman Holly Patterson who died as a result of taking RU486 in 2003 "These are violently active chemicals and they have violent reactions on the organism … [What is the] situation in which a woman would undergo that kind of assault?" – Australian feminist Dr Germaine Greer, addressing gynaecologists and obstetricians in 2002 "A drug which ends a new human life and endangers a woman's health is never a ‘safe and effective' solution." – Dr Brigid Vout, Life Office, Catholic Diocese of Sydney What is RU486? RU486 is not the same as the "morning after" pill (Postinor-2). RU486 is the generic term for mifepristone, an artificial steroid that blocks progesterone, a vital nutrient hormone. It causes the nutrient lining of the mother's uterus to disintegrate, and the embryo withers and dies. A second drug, misoprostol, a prostaglandin developed to treat ulcers, is used 48 hours later to induce uterine contractions that detach and expel the embryo and uterine contents. More than one million women worldwide have used RU486 to end their pregnancy. RU486 is effective from the fifth to the seventh week following the last menstrual period, with decreasing effectiveness up to the ninth week. Used alone, RU486 has an abortion rate of 60-80 per cent. Used with misoprostol, this rises to 95 per cent. Mifepristone is also used to treat certain rare forms of cancer, and may have other therapeutic applications. Mifepristone was developed by Roussel-Uclaf, a French pharmaceutical company. Possible side effects and complications A common side-effect is severe pain similar to that of miscarriage, with over half of women needing specific pain medication and one-third needing narcotics. Other side-effects may include nausea and dizziness, syncope (brief loss of consciousness), serious bacterial infection, sepsis, prolonged bleeding (averaging from 9 to 30 days) and death. Some women who experienced severe bleeding as a result of taking the drug required blood transfusions. Women lacking ready access to ultrasound and blood transfusion, such as those in remote communities and developing countries, are more likely to die. The drug does not affect ectopic
JUEVES 12 DE ABRIL DEL 2012 JUEVES, 12 DE ABRIL DEL 2012 EE.UU. acusa a Apple y a cinco Nokia, fabrican- n Volkswagen y editoriales de manipular precios te finlandés de BMW, automotrices teléfonos móviles, alemanas, registra- advirtió que sus ventas ron nuevos récords fueron más débiles



