



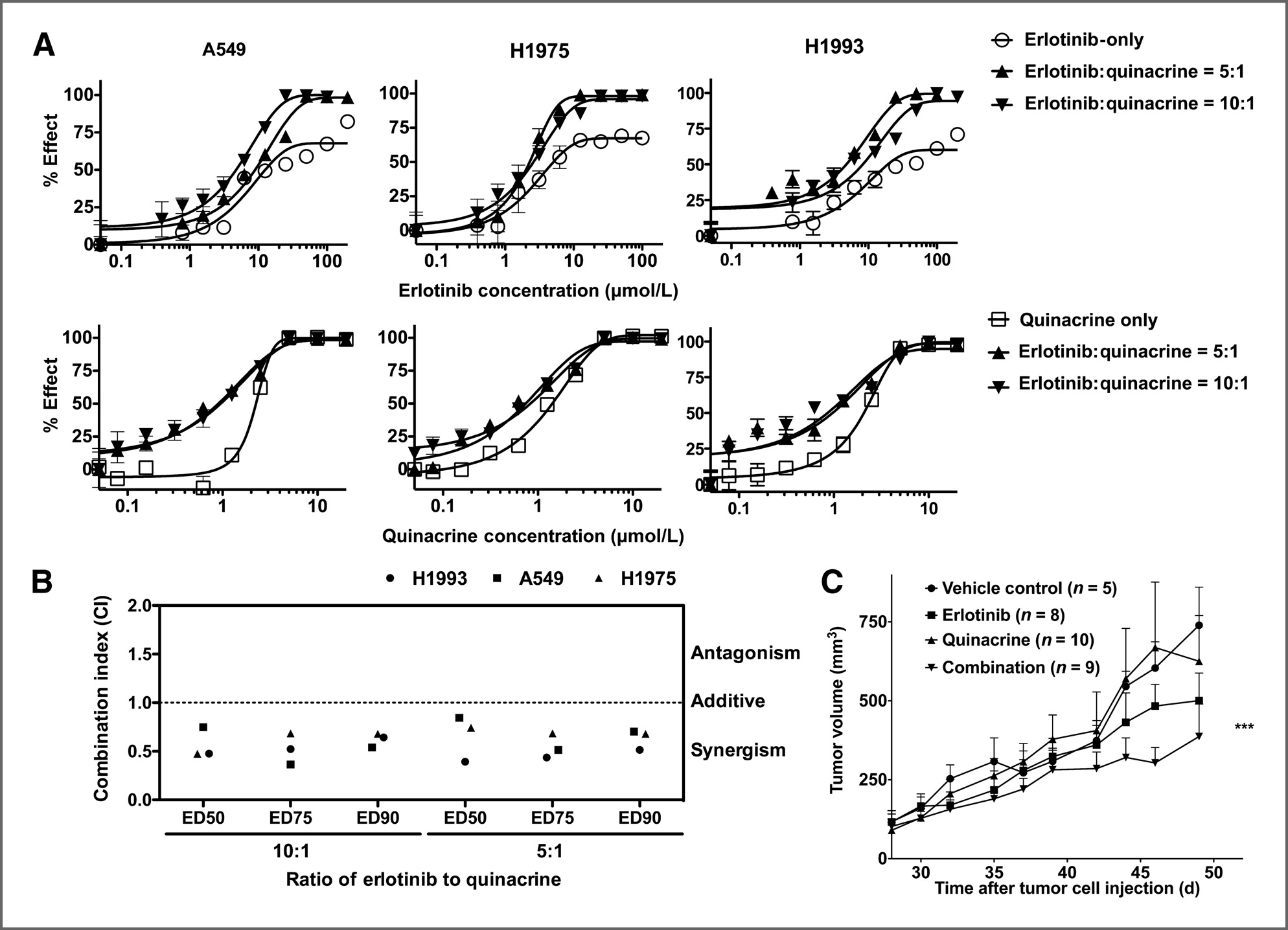
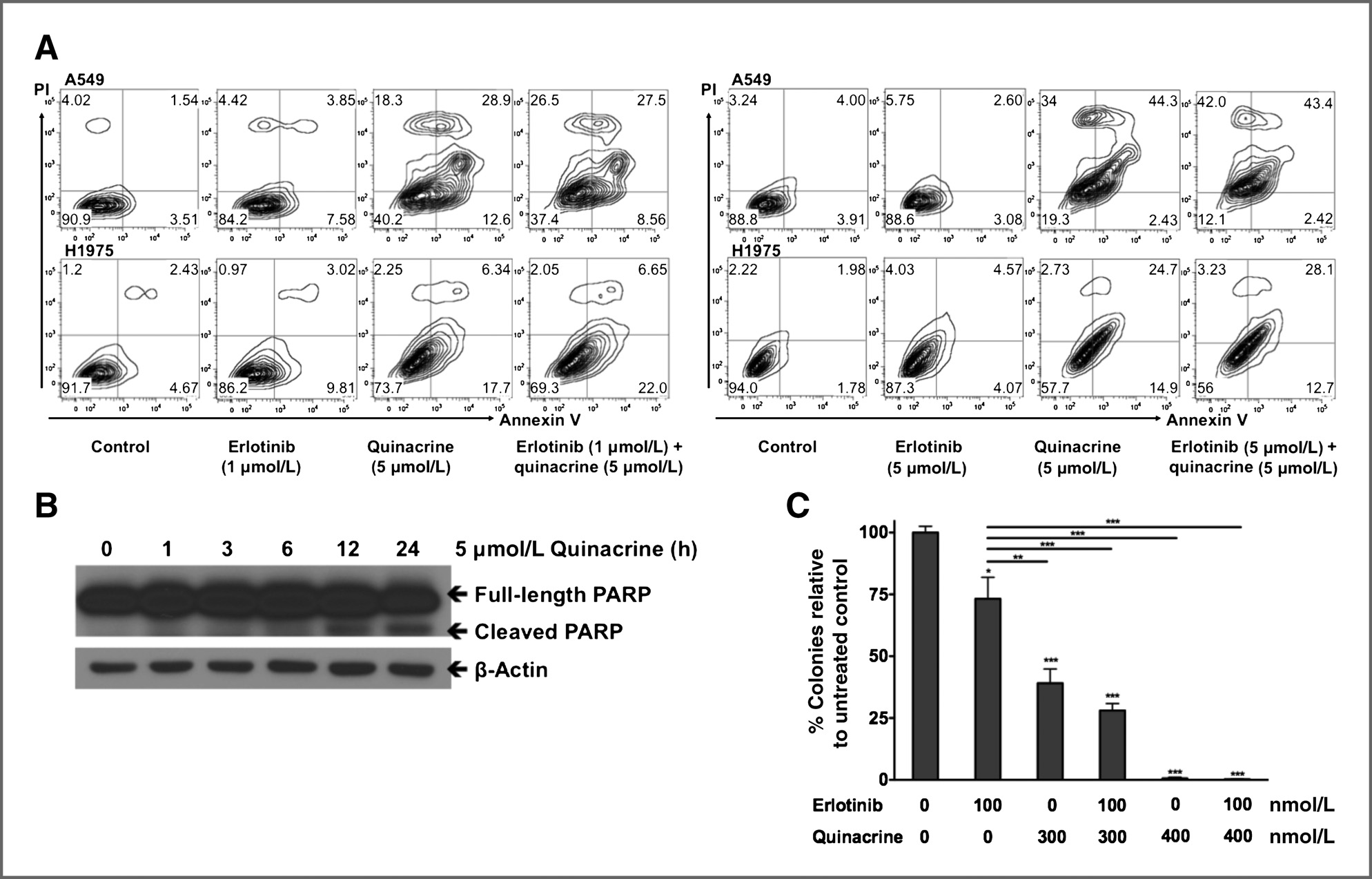
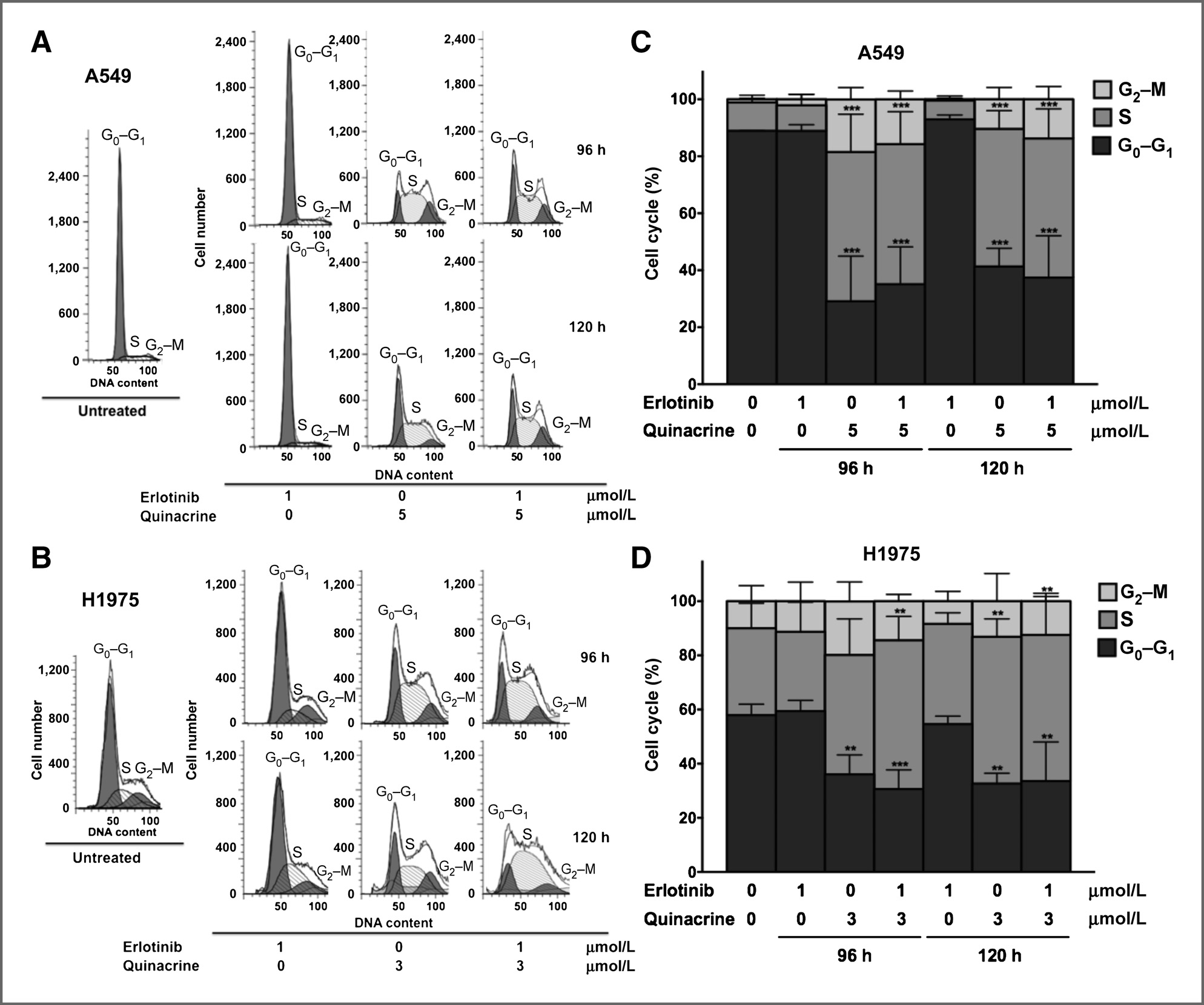
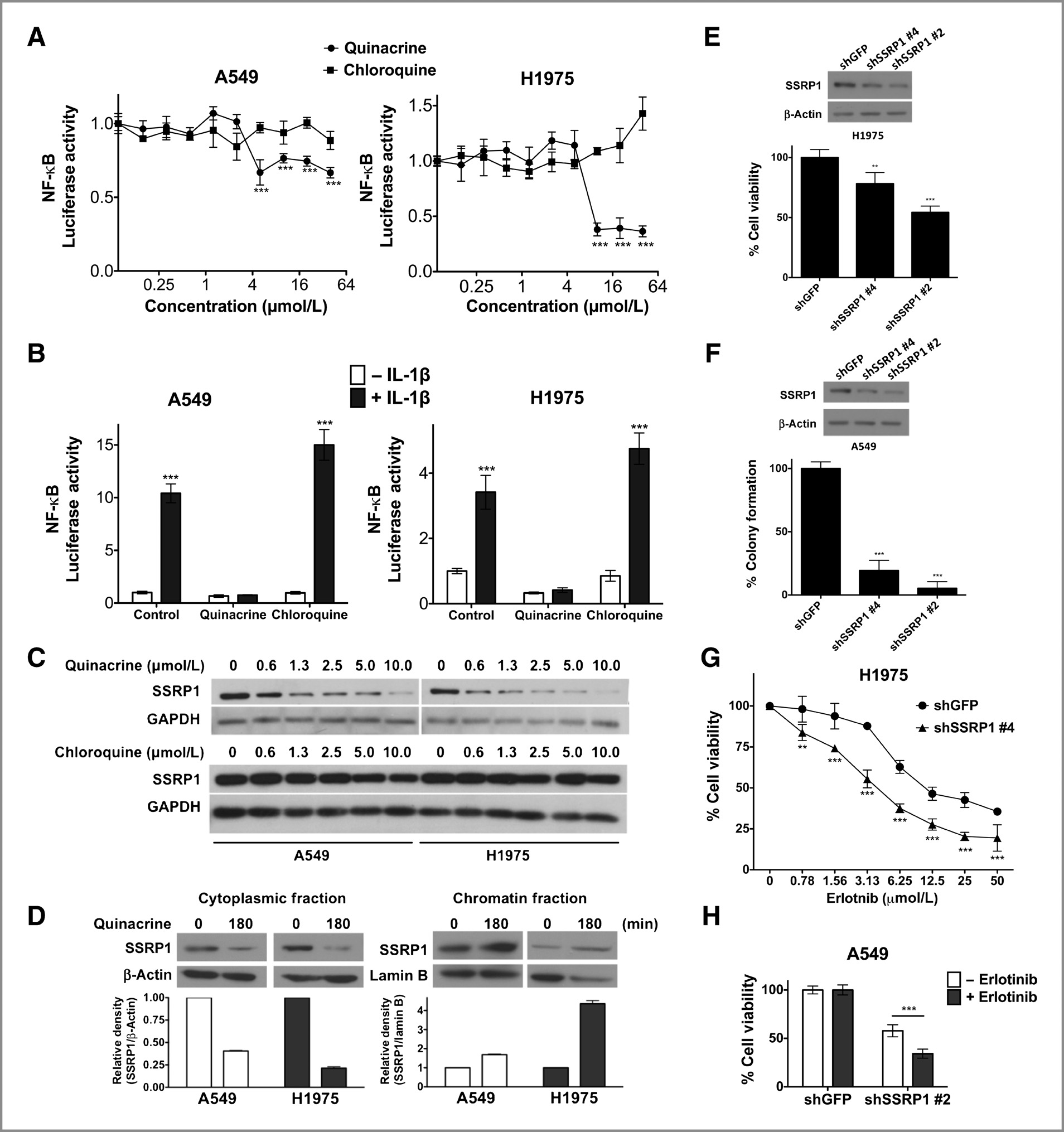
Isc310343.qxd
and Developing Academic and Behavioral Interventions for Students with Bipolar KIM KILLU AND R. MARK A. CRUNDWELL Despite significant advances in practices for effectively designing anddelivering instruction for students with disabilities, educators continueto face challenges addressing the needs of students with emotionaland behavioral disorders. Little information is available for educatorson accommodations and modifications that would serve the needs ofthese students and address the unique challenges they present in theclassroom. The educational, social, and behavioral needs of studentswith bipolar disorder are discussed along with suggestions for provid-ing effective accommodations and modifications in the classroom.