Vitamin e, vitamin c und provitamin a
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
Aktueller Forschungsstand zur neuroprotektiven Therapie der
Von Dr.med. Herwig W. Lange
Da ich vom Sozialgericht in Detmold zum Gutachter bestellt worden bin zur Frage, welche Vitamine und Naturstoffe mit Aussicht auf Erfolg bei Huntington-Kranken eingesetzt werden können, habe ich mir noch mal die Mühe gemacht, alle relavanten Publikationen zu dem Thema zu durchforsten. Die Quintessenz möchte ich auf diesem Wege allen Interessierten zugänglich machen. Die hier enthaltene Information darf gerne zitiert und verwendet werden, aber bitte mit Quellenangabe. Den meisten Mitgliedern der DHH und Lesern des Huntington-Kuriers ist bekannt, dass ich vom Beginn meiner Tätigkeit für Huntington-Kranke im Jahr 1980 immer nach besseren Behandlungs-möglichkeiten gesucht habe – nicht nur nach besser wirksamen und verträglicheren Medikamenten für die jeweiligen Symptome der Huntington-Krankheit (HK), sondern auch nach Wegen, das Fortschreiten der Erkrankung wenigstens zu bremsen. Gleichzeitig war mir von Anfang an wichtig, die negativen Auswirkungen der HK auf Patienten und Familienangehörige möglichst gering zu halten und die Lebensqualität aller Betroffenen zu verbessern. Dabei lernte ich sehr schnell, welch mächtigen Einfluss die Lebensumstände der Huntington-Kranken auf die Lebensqualität und auch auf den Verlauf der Krankheit haben, z.B. dass eineiige Zwillinge mit der HK nicht am selben Tag erkranken (es lagen in einem Fall mehr als 5 Jahre zwischen den Manifestationszeitpunkten) und dass bei Geschwistern bis zu 25 Jahre Unterschied im Manifestationsalter zu finden waren, wobei die Geschwister mit den besseren Lebensumständen regelmäßig den späteren Krankheitsbeginn und den milderen Verlauf aufwiesen. Durch meine engen Kontakte mit der Arbeitsgruppe von Susan Folstein am Johns Hopkins Hospital in Baltimore, MD, USA, erfuhr ich Ende der 80er Jahre, dass die Arbeitsgruppe die neuroprotektive Wirkung von Vitamin E und C als Antioxidantien bei Huntington-Kranken untersuchte und von deren positiver Wirkung auf einen Teil der Patienten, nämlich solchen mit noch geringen neurologischen Störungen. Seit der Erörterung dieser Ergebnisse habe ich Huntington-Patienten und -Risikoperson seit 1990 die tägliche Einnahme von Vitamin E in Kombination mit Vitamin C und ß-Carotin (Provitamin A), bei dem die Gefahr einer Überdosierung wie bei Vitamin A nicht besteht, empfohlen. Mit den Fortschritten der wissenschaftlichen Erkenntnisse kamen dann die Substanzen Coenzym Q10, Kreatin und zuletzt Ethyl-
Eicosapentaensäure (EPA) hinzu. Einen ähnlichen Therapieansatz verfolgen seit einigen Jahren GOODMAN & GOODMAN in den USA mit dem Huntington's Disease Drug Works (s. SCIENCE Vol. 304, 7 May 2004, 816-817): sie empfehlen ihren Huntington-Patienten Blaubeeren-Extrakt, Coenzym Q10, Kreatin, Cysteamin,
Omega-3-Fettsäuren und Trehalose (die bei Huntington-Mäusen und –Ratten neuroprotektiv wirkt, aber beim Menschen nicht wirken kann, da die Trehalose beim Menschen in der Darmwand in 2 Glukose-Moleküle gespalten wird). Auch bei anderen neurodegenerativen Erkrankungen wie z.B. Morbus Alzheimer oder Parkinson oder beim Alterungsprozess spielt der oxidative Stress offensichtlich eine pathogenetische Rolle, wie dem Buch „Oxidativer Stress und Pharmaka", herausgegeben von SIEMS, KRÄMER und GRUNE (2005) zu entnehmen ist: „
Eine antioxidative Therapie von neurodegenerativen Krankheiten könnte durchaus viel verspre-chend sein, gerade wenn über lange Zeit hinweg therapiert werden kann und vor allem bereits in präklinischen Stadien damit begonnen wird. Dazu wird eine Kombination verschiedener Antioxi-dantien notwendig sein, die wahrscheinlich in klar definierten Zeitfenstern zur Anwendung kommen muss. Die antioxidative Therapie neurologischer Krankheiten steht derzeit wohl erst ganz am Be-ginn ihrer Möglichkeiten." Hauptangriffspunkt dieser Komplex-Therapie ist der oxidative Stress, der bei der HK – wie auch bei der Alzheimer- oder Parkinson-Krankheit – eine entscheidende Rolle im Krankheitsprozess
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
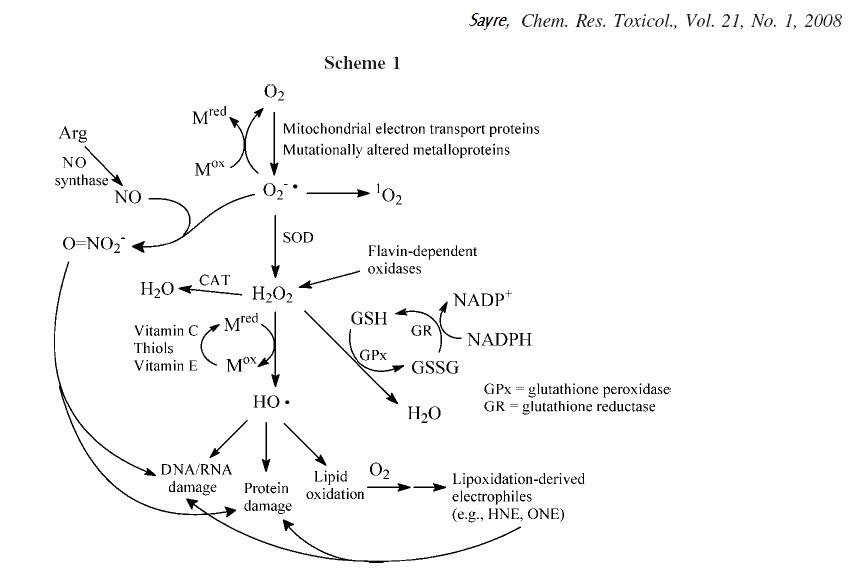
spielt. Abbildung 1 zeigt schematisch die Bildung der freien Radikale und ihre schädlichen Auswirkungen auf proteine, Lipide und die DNS (Erbsubstanz). Abbildung 1:
Die HK ist im Tierexperiment in ihrem Verlauf beeinflussbar, wie zahlreiche Untersuchungen an der Huntington-Maus belegen. Neben verschiedenen Substanzen, u. a. Coenzym Q10 und Kreatin,
hat sich auch eine anregende Umgebung als neuroprotektiv wirksam erwiesen (VAN DELLEN et al., 2000; HOCKLY et al., 2002; GLASS et al., 2004). Umwelteinflüsse sind auch bei Huntington-Kranken von Bedeutung. Eineiige Zwillinge mit der HK haben durchaus keinen identischen Krankheitsverlauf, wie ich aus eigener klinischer Erfahrung weiß und von verschiedenen Autoren publiziert wurde. Dieses „proof of principle" belegt, dass Umweltfaktoren den Verlauf der HK beeinflussen können, wie das auch aus den Untersuchungen am riesigen Huntington-Stammbaum am Lake Maracaibo in Venezuela hervorgeht (WEXLER et al., 2004). Die Mehrzahl der Huntington-Experten geht davon aus, dass ein multimodaler Therapieansatz, der die bisher bekannten, komplexen zerebralen Stoffwechselstörungen im Gefolge der HK berücksichtigt, zur Zeit am aussichtsreichsten ist. Die von mir schon im letzten Jahr im Internet und im Huntington-Kurier aufgeführten wissenschaftlichen Untersuchungen an Huntington-Kranken mit Vitaminen und ähnlichen Substanzen belegen, dass eine positive Beeinflussung des Verlaufs der HK mit diesen Substanzen möglich ist. Als Einzelsubstanzen werden jetzt Coenzym Q10 und Kreatin neben bekannten Medikamenten und
neuen Substanzen in weiteren wissenschaftlichen Studien auf ihre Wirksamkeit bei Huntington-Kranken und z.T auch bei Risikopersonen untersucht. Es kann den Huntington-Kranken aber nicht zugemutet werden, bis zum Eintreffen weiterer Daten aus Therapiestudien der drohenden Progredienz ohne Behandlung ausgeliefert zu sein. Durch die weiter unten genannten Naturstoffe lässt sich mit einem hohen Grad an Wahrscheinlichkeit der Verlauf der HK spürbar positiv beeinflussen. Diese Substanzen werden in der präklinischen und der frühen klinischen Phase besser wirken als in der Endphase der HK. Ihre Wirkung wird nicht ausreichen, um das Auftreten der HK bei Gen-tragenden Risikopersonen zu verhindern, sie sind
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
aber wahrscheinlich geeignet, die Manifestation hinauszuzögern. Bei Huntington-Kranken konnten mit diesen Substanzen in mehreren wissenschaftlichen Untersuchungen (s. Webseite der DHH und Huntington-Kurier) sowohl relevante Stoffwechselparameter als auch der klinische Verlauf positiv beeinflusst werden.
Zu den von der Kägerin eingenommenen Substanzen:
Vitamin E, Vitamin C und (Pro)Vitamin A
Bezug zur HK: Reduktion von oxidativem Stress.
Vitamin E (Tocopherol) ist ein wichtiges Antioxidans, es sitzt in der Zellmembran und schützt die
Lipide der Zellmembranen vor der Oxidation durch freie Radikale und Sauerstoffradikale. Bei Vita-
min E ist eine moderate Hochdosierung bis 270 mg, entsprechend ca. 400 Internationalen
Einheiten (IE), nach Einschätzung der EFSA gesundheitlich unbedenklich, bei höheren Dosen (>
400 IE) haben epidemiologische Studien in jüngster Zeit eine leicht erhöhte kardiovaskläre
Sterblichkeit gefunden, wobei die wissenschaftliche Diskussion zu diesen Daten noch im Gange
ist. γ-Tocopherol (überwiegt in der Nahrung) kann in Tumorzellen die Apoptose auslösen, was die
Gefahr birgt, dass das Absterben der gefährdeten Neurone bei der HK gefördert wird. Daher ist die
Gabe von α-Tocopherol unbedingt vorzuziehen.
Für seine Wirkung ist Vitamin E auf Vitamin C (Ascorbinsäure) angewiesen, das das verbrauchte,
oxidierte Vitamin E wieder reduziert. Vitamin C findet sich in den Körperflüssigkeiten sowie Zytop-
lasma und kann Sauerstoffradikale entschärfen. Bei Vitamin C ist eine Überdosierung nicht mög-
lich, in seinem bekannten Selbstversuch konsumierte Linus Pauling im Alter täglich 18 g Vitamin C
und erreichte ein Lebensalter von 93 Jahren. Überflüssiges Vitamin C wird rasch renal ausge-
schieden.
Provitamin A (ß-Carotin) wird im Organismus bedarfsgerecht zu Vitamin A umgewandelt. Es ist
das wirksamste Mittel zur Entschärfung des giftigen Singulett-Sauerstoffs. Eine Überdosierung
durch Höchstdosen ist möglich, wahrscheinlich bedingt durch toxische Abbauprodukte. Raucher
sollten wegen Erhöhung des Bronchialkrebsrisikos kein ß-Carotin einnehmen. Eine tägliche Zufuhr
von bis zu 10 mg hält die SCF der EU-Kommission für unbenklich, das Bundesinstitut für
gesundheitlichen Verbraucherschutz und Veterinärmedizin (BgVV) empfiehlt 2 mg ß-Carotin pro
Tag in isolierter Form als Höchstmenge. Nach SIEMS et al. (2005) sind Supplementdosen bis 6
mg/Tag sicher. Zur Optimierung der Antioxidation soll gemäß DGE eine Plasmakonzentration von
> 0,4 µmol/l erreicht werden.
Vitamin A (Retinol) hat beim erwachsenen Menschen eine Wirkung auf Haut und Schleimhäute
sowie die Retina, es hat einen Einfluss auf die Genexpression. Ob letzterer Effekt bei Huntington-
Kranken therapeutisch genutzt werden kann, ist unbekannt. Ob es selbst als Antioxidans wirkt, ist
umstritten. In Zelkulturen hat Retinol den oxidativen Stress erhöht.
Es führt bei akuter Überdosierung zu Kopfschmerzen, Müdigkeit, Übelkeit, Papillenödem, bei
chronischer Überdosierung zu Hautveränderungen, Knochenschmerzen, Müdigkeit und Blutungen,
später zu Leberfibrose und Pseudotumor cerebri durch Erhöhung des Liquordruckes.
Untersuchungen bei Huntington-Patienten
Die Gruppe um S. FOLSTEIN am Johns Hopkins Hospital in Baltimore, MD, USA, (PEYSER et al.,
1995) untersuchte Ende der 80er Jahre die neuroprotektive Wirkung von Vitamin E und C als
Antioxidantien. In einem Doppelblind-Versuch erhielten 81 Patienten für 1 Jahr 3x 1.000 IE d-α-
Tocopherol bzw. Plazebo, alle Patienten bekamen 2x 0,5 g Vitamin C und 2x 25.000 IE Vitamin A,
das gegeben wurde, um einem möglichen negativen Einfluss der hohen Vitamin-E-Gabe auf den
Vitamin-A-Metabolismus zu vermeiden. 7 Patienten beendeten die Studie nicht, auswertbar waren
die Daten von 73 Patienten. Insgesamt waren die 40 Verum-Patienten nach 1 Jahr weder im
quantitativen neurologischen Befund (QNE), noch im Mini-mental-State, noch bei den Alltags-
aktivitäten besser als die 33 Placebo-Patienten, bei denen sich möglicherweise die Gabe von
Vitamin C und A positiv ausgewirkt hat. Bei den weniger betroffenen Patienten (QNE <45) ergab
sich eine signifikante Reduktion der neurologischen Symptome. Dieses Ergebnis kann als Hinweis
auf eine neuroprotektive Wirkung von Vitamin E im Zusammenspiel mit Vitamin C und A in der
frühen Phase der HK gewertet werden.
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
Coenzym Q10
Bezug zur HK: Mitochondrien-Funktion und oxidativer Stress. Q10 ist notwendig für die Funktion der Atmungskette in den Mitochondrien und wirkt als Radi-
kalenfänger antioxidativ. Es wird vom menschlichen Organismus selbst synthetisiert, was aber bei Huntington-Kranken offensichtlich nicht ausreicht. Bei unbehandelten Huntington-Patienten ist Q10
im Serum erniedrigt und lässt sich durch orale Gabe normalisieren, wie die Bochumer Arbeitsgruppe (ANDRICH et al., 2004) nachgewiesen hat. Bei Parkinson-Kranken konnte gezeigt werden, dass eine Nano-Präparation von Q10 in der Dosierung von 300 mg/d sicher und gut
verträglich ist sowie zu Plasmaspiegeln führt, die Q10-Standardformulierungen von 1200 mg/d
ähnlich sind. Untersuchungen bei Huntington-Patienten Im Gehirn von Huntington-Kranken sind erhöhte Laktat -Spiegel in Hirnrinde und Striatum in einigen Studien nachgewiesen, sie ließen sich mit Q10 senken (KOROSHETZ et al. 1997).
In der CARE-HD-Studie waren die mit Q10 behandelten Patienten am Ende der 30-monatigen
Beobachtungszeit 13 % funktionsfähiger in den Alltagsaktivitäten als die Patienten unter Placebo oder Remacemid. Leider war die Studie von den Fallzahlen so angelegt, dass erst eine Differenz von 40 (!) % signifikant gewesen wäre. In der Pre-2CARE-Studie untersuchte die HUNTINGTON STUDY GROUP (HSGN, 2007) die Sicherheit und Verträglichkeit von Q10 in einem offenen Behandlungsversuch über 20 Wochen bei 20
Huntington-Kranken und 8 gesunden Kontrollen. Alle Probanden erhielten zunächst 1200 mg/d Q10, nach 4 Wochen wurde die Dosierung erhöht auf 2400 mg/d und nach 8 Wochen auf 3600
mg/d. Die Probanden wurden dann alle 4 bis maximal 20 Wochen im Serum auf 8-Hydroxy-2'-Deoxyguanosin (8OH2'dG), als Marker für oxidative Schäden an der DNS, und Q10 untersucht.
Sowohl bei den Kontrollpersonen als auch bei Huntington-Kranken war 8OH2'dG am Ende der Behandlung signifikant reduziert. Es wird derzeit eine neue Untersuchung mit hohen Q10-Dosen (1,2 und 2,4 g/d) in Nordamerika
durchgeführt [Massachusetts General Hospital in Zusammenarbeit mit der University of Rochester, gefördert vom National Institute of Neurological Disorders and Stroke (NINDS)]. Eine weitere Studie (PREQUEL) untersucht die Verträglichkeit und biologische Aktivität von Coen-zym Q10 in präklinischen HD-Genträgern, gefördert vom NINDS.
Nebenwirkungen einer langfristigen, auch hoch dosierten Gabe sind nicht bekannt, jedoch bedarf die Therapie bei Herzkranken einer engmaschigen Überwachung. Wegen der besseren Verträglichkeit und Bioverfügbarkeit sollte Coenzym Q10 als Nanopräparation gegeben werden.
Ethyl-Eicosapentaensäure (EPA)
Bezug zur HK: schützt Mitochondrien, hemmt Phospholipasen sowie Caspasen und so die
Apoptose, stabilisiert Zellmembranen.
EPA wird vom menschlichen Organismus selbst synthetisiert, bei Huntington-Kranken besteht aber
offensichtlich ein erhöhter Bedarf.
Untersuchungen bei Huntington-Patienten
VADDADI et al. (2002) behandelten 17 Huntington-Kranke über 19 Monate mit 2x 1 Kapsel, die ein
Gemisch aus 70 mg γ-Linolensäure, 35 mg EPA, 20 mg Docosahexaensäure, 50 mg α-Liponsäure
und 30 mg d-α-Tocopherol enthielt. Am Ende der Studie waren alle Verum-Patienten auf der
Unified Huntington's Disease Rating Scale (UHDRS) und der Rockland-Simpson Dyskinesia
Rating Scale (RSDRS) – in letzterer signifikant - verbessert, während sich alle Patienten mit
Placebo auf diesen Skalen verschlechterten. Hier ist nicht zu entscheiden, welche der protektiven
Substanzen den Ausschlag gab, möglicherweise war das Gemisch per se für die Wirksamkeit
verantwortlich.
In einer kleinen Studie mit 7 Heimpatienten im fortgeschrittenen Stadium (III) der HK (PURI et al.,
2002) erhielten 3 Patienten EPA, 4 Placebo. Nach 6 Monaten zeigten sich alle mit EPA
behandelten Patienten auf der orofazialen Subskala verbessert, während sich alle Patienten mit
Placebo auf dieser Skala verschlechterten. Der Unterschied der Gruppen war signifikant.
In einer weiteren Studie (PURI et al., 2005) an 135 Huntington-Kranken mit weniger starken
Symptomen ergab die Behandlung mit EPA 2 g/d bei der Subgruppe der 83 Patienten, die über die
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
ganze zweijährige Untersuchungszeit beobachtet werden konnten, einen positiven Effekt auf die Motorik, besonders deutlich bei Patienten mit weniger als 51 CAG. Bei einer neuerlichen Doppelblind-Untersuchung mit EPA in Europa (290 Patienten) und Nord-amerika (316 Patienten) war nach 6-monatiger Behandlungszeit der Effekt von 2 g/d EPA auf die Motorik nicht stärker als der von Placebo (HSGN, 2007, Pressemitteilung). Die Untersuchung wurde in Nordamerika um 6 Monate verlängert, in dieser Zeit erhielten alle Patienten EPA. Am Ende der Untersuchung waren die Patienten, die EPA über die gesamten 12 Monate erhalten hatten, in der Motorik signifikant besser (keine Veränderung in der Motorik gegenüber einer Verschlechterung um 1,8 Punkte bei dem Patienten, die EPA nur 6 Monate erhalten hatten (p = 0,02), besonders deutlich war der positive Effekt für die Patienten mit weniger als 45 CAG (Verbesserung um 1,2 Punkte gegenüber einer Verschlechterung um 1,6 Punkte (p = 0,004). Nebenwirkungen einer langfristigen, auch hoch dosierten Gabe sind nicht bekannt. Ein Vorteil der (teureren) rezeptpflichtigen Präparate gegenüber den Nahrungsergänzungsmittel-Präparaten ist nicht erkennbar, letztere gibt es als 1-g-Kapseln mit 18 % EPA und 12 % Docosahexaensäure (DHA). DHA wird vom Huntington's Diesease Drug Works (http://hddrugworks.org) favorisiert we-gen der günstigen klinischen Resultate bei Alzheimer-Patienten. Da die Frage EPA versus DHA wissenschaftlich noch nicht abschließend beantwortet werden kann, ist eine Kombinationstherapie mit EPA und DHA am aussichtsreichsten.
Kreatin
Bezug zur HK: intrazellulär Energiespeicher, Mitochondrienschutz und Antioxidans.
Kreatin wird durch die Kreatinkinase in Phosphokreatin umgewandelt, das aus ADP ATP, den
wichtigsten Energielieferanten für alle chemischen Prozesse im Organismus, generieren kann. Es
wird vom menschlichen Organismus selbst synthetisiert, bei Huntington-Kranken besteht aber
wahrscheinlich ein Mangel. Die maximale Rate der mitochondrialen ATP-Synthese ist in Muskeln
von Huntington-Kranken verringert, auch bei präsymptomatischen Risikopersonen (LODI et al.,
2000). SÁNCHEZ–PERNAUTE et al. (1999) fanden in den Basalganglien von Huntington-Patienten
eine 60-prozentige Reduktion von Kreatin, bei Risikopersonen eine Abnahme um 30 %.
Untersuchungen bei Huntington-Patienten
HERSCH und Mitarbeiter (2006) konnten in einem Doppelblind-Versuch mit 64 Huntington-
Patienten zeigen, dass 8 g/d Kreatin über 16 Wochen gut vertragen wurde. Die Kreatinkon-
zentration im Serum und Gehirn erhöhten sich in der Kreatin-behandelten Gruppe und gingen nach
Absetzen zurück auf den Ausgangswert. Im Serum war 8-hydroxy-2'-deoxyguanosin (8-OH2'dG)
bei Huntington-Kranken deutlich erhöht und wurde durch die Kreatinbehandlung verringert.
In 2 Untersuchungen (TABRIZI et al., 2005 und VERBESSEM et al., 2003) zeigten die mit Kreatin
behandelten Huntington-Kranken nach 12 Monaten keine Verschlechterung in Motorik- oder
neuropsychologischen Tests. Bei ATL-Funktionen waren die Ergebnisse uneinheitlich: Die Gruppe
um Tabrizi sah keine Verschlechterung, während die Gruppe um Verbessem eine Abnahme der
ATL-Funktionen feststellte.
Später führte die Arbeitsgruppe von HERSCH (2007) eine offene Dosisfindungsstudie an 10
Huntington-Patienten mit frühen Symptomen durch. Sie ermittelte 30 g Kreatin täglich als die
optimale Dosis, basiernd auf dem höchsten erreichbaren Plasmaspiegel, der mit MRS ge-
messenen Zunahme im Gehirn, der verringerten Verträglichkeit bei noch höheren Dosen, sowie
der dosisabhängigen Reduktion des 8-OH2'dG auf den Normalwert. Auch der kognitive Abbau war
verlangsamt und die Geschwindigkeit der regionalen Gehirnatrophie, ermittelt durch MRT-
Morphometrie, verringert.
Kreatin wird jetzt auch bei Risikopersonen in einer Studie (PRECREST) am Massachusetts Gene-
ral Hospital untersucht.
Diese Befunde belegen, dass Kreatin in hohen Dosen verträglich ist und den Krankheitsverlauf
positiv verändern kann. An Nebenwirkungen sind bekannt: häufig Gewichtszunahme (1 - 5 kg),
überwiegend bedingt durch Wassereinlagerung in der Muskulatur, Übelkeit, Erbrechen und
Diarrhöe, selten Muskelkrämpfe, Leber- und Nierenfunktionsstörungen (PERSKY et al., 2001).
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
Vita Lecithin
Zusammensetzung (Deklarationen des Herstellers): 1 Softgel enthält 1,2 g Soja-Lecithin mit min-
destens 60% Phospholipiden, u. a. Cholin, Inositol und Linolsäure.
Bezug zur HK: Cholinerges Defizit und Schutz der Zellmembranen.
LANGE & THÖRNER (1975) konnten einen 50-%igen Verlust der cholinergen Interneurone im Stria-
tum nachweisen. Post mortem zeigt sich eine Reduktion von Acetylcholin im Striatum. MANYAM et
al. (1990) wiesen einen reduzierten Cholin-Spiel im Liquor nach.
GÓMEZ-ANSÓN et al. (2007) fanden bei 17 präklinischen Huntington-Personen eine Reduktion von
Cholin im frontalen Kortex und vermuteten einen Zusammenhang mit den gleichfalls festgestellten
neuropsychologischen Defiziten.
Untersuchungen bei Huntington-Patienten
Die Arbeitsgruppe um DAVIS untersuchte Anfang der 70er Jahre die Wirkung von Cholin auf Pati-
enten mit Spätdyskinesien oder HK, veröffentlicht wurden jedoch nur vorläufigen Daten.
BARBEAU schrieb 1978 einen Übersichtsartikel zur Therapie neurologischer Krankheiten mit oraler
Zufuhr der Acetylcholin-Vorläufer Cholin oder Lecithin zwecks Korrektur von insuffizienten choli-
nergen Systemen im Gehirn. In seine Überlegungen bezog er die HK, das Gilles-de-la-Tourette-
Syndrom, die Friedreich-Ataxie und die präsenile Demenz ein. Vorläufige Daten verschiedener
Arbeitsgruppen zeigten seiner Meinung nach, dass bei jeder der genannten Krankheiten gelegent-
lich eine gewisse klinische Verbesserung zu sehen war. Barbeau schlug weitere Untersuchungen
mit dieser Fragestellung vor.
Weitere Untersuchungen an Huntington-Kranken sind nicht publiziert.
Aufgrund dieser Datenlage erscheint es fraglich, ob über den Verzehr von z.B. täglich 1 Ei hinaus
die zusätzliche Zufuhr von Lecithin einen therapeutischen Nutzen bei der HK hat.
Vit-Amino (Aminosäuren)
Zusammensetzung (Herstellerangaben): pro Kapsel L-Arginin (Hydrochlorid) 50 mg,
L-Cystein (Hydrochlorid) 50 mg, L- Histidin 50 mg, L-Isoleucin 50 mg, L-Leucin 50 mg, L-Lysin
(Hydrochlorid) 50 mg, L-Methionin 50 mg, L-Ornithin (Hydrochlorid) 50 mg,
L-Phenylalanin 50 mg, L-Threonin 50 mg, L-Tyrosin 50 mg, L-Valin 50 mg.
Bezug zur HK: gestörte Balance von Aminosäuren im Gehirn.
OEPEN et al. (1982) fanden eine Reduktion von Asparagin, Isoleucin, Leucin, Phenylalanin, Histi-
din, Aarginin, α-Aminoadipin-Säure and Homocarnosin im Liquor von Huntington-Patienten. Im
Plasma von Huntington-Kranken wurden von unterschiedlichen Gruppen (PERRY et al. 1969 und
1972, WATT et al. 1978, REILMANN et al. 1995, UNDERWOOD 2006, MOCHEL et al., 2007) unter-
schiedliche Veränderungen der Aminosäuren beschrieben.
Untersuchungen bei Huntington-Patienten:
MCLEOD & DE L. HORNE (1972) behandelten 4 Huntington-Patienten für 14 Tage mit 7 g L-
Tryptophan und 70 mg Pyridoxin und fanden keine Veränderung in der Motorik. BARR et al. (1978)
behandelten 5 Huntington-Kranke über 2 Jahre mit 25 g L-Glutamat und 500 mg Pyridoxin, sie
sahen weder in der Motorik noch im Verhalten eine Verbesserung.
Hühnereier haben einen hohen Gehalt an essentiellen Aminosäuren, 1 Hühnerei pro Tag deckt
den Bedarf bei gesunden Erwachsenen weitgehend. Gründe für eine zusätzliche Gabe von essen-
tiellen Aminosäuren zur Behandlung von Huntington-Kranken sind nicht erkennbar.
Taurin
Bezug zur HK: Oxidativer Stress und GABA-Mangel.
Taurin, eine Aminosulfonsäure, hat antioxidative Eigenschaften und wirkt GABA-A-agonistisch.
Neue Untersuchungen weisen darauf hin, dass die zytoprotektiven Wirkungen sowohl den Effekten
von Chaperonen als auch denen von Antioxidantien ähneln und sie für den Alterungsprozess we-
sentliche zelluläre Funktionen beeinflussen, z. B. die Apoptose oder die Aufrechterhaltung der mi-
tochondrialen Integrität. Welche Rolle dem Taurin im Alterungsprozess oder bei Krankheiten, die
mit oxidativen Stress einhergehen, zukommt, ist Gegenstand der Forschung, z.B. im Rahmen des
von der Deutschen Forschungsgemeinschaft geförderten Graduiertenkolleges "Molekulare Ziele
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
von Alterungsprozessen und Ansatzpunkte der Alterungsprävention" an der Heinrich-Heine-Universität Düsseldorf. Ein Dissertationsprojekt beschäftigt sich mit der Aufklärung der protektiven Wirkung von Taurin. ELLISON et al. (1987) fanden post mortem in Gehirnen von Huntington-Patienten in 9 kortikalen und 9 subkortikalen Hirnregionen keine Unterschiede im Tauringehalt im Vergleich zu Kontrollgehirnen, BONILLA et al. (1988) eine Erhöhung von Taurin im Brodmann-Areal 10 (lokalisiert am Frontalpol des Gehirns). Untersuchungen bei Huntington-Patienten: keine veröffentlicht. NYLAND et al. (1989) untersuchten 14 Patienten mit Dyskinesien. Die Patientin erhielten 3 g/d Tau-rin oral. 6 Patienten zeigten initial eine Verbesserung, die jedoch nicht bis zum Ende der 6-wöchigen Behandlungsperiode anhielt. Der erwachsene menschliche Organismus kann Taurin aus der Aminosäure Cystein selbst herstel-len. Eine Zufuhr durch Nahrungsmittel ist daher bei gesunden Erwachsenen nicht nötig. Ob Hun-tington-Kranke von einer zusätzlichen Gabe profitieren, kann beim derzeitigen Kenntnisstand wis-senschaftlich nicht abschließend beurteilt werden. Ein schädlicher Einfluss ist eher unwahrschein-lich, ein Nutzen theoretisch denkbar durch die Schutzfunktion bei den Mitochondrien.
Superoxid-Dismutase (SOD)
Bezug zur HK: Radikalfänger.
SOD ist in nahezu allen Körperzellen vorhanden, es liegt einmal als zytosolisches Enzym Cu/Zn-
Superoxid-Dismutase (SOD1) vor, zum anderen als mitochondriale Mn-Superoxid-Dismutase
(SOD2). Beim Menschen ist auch eine extrazelluläre Form bekannt (SOD3). Etwa 2 – 5 % des
vom Gehirn aufgenommenen O2 wird in Superoxidanion-Radikale umgewandelt, welche durch die
SOD in das weniger gefährliche H2O2 umgesetzt werden.
Während oral zugeführte SOD im Magen/Darmtrakt degradiert wird, konnten MUTH et al. (2004) zeigen, dass pflanzliche SOD, gebunden an Gliadin, bei oraler Zufuhr resorbiert wird und bei Ver-suchspersonen die DNA in Leukozyten vor dem oxidativen Stress einer hyperbaren Sauerstoffbe-handlung schützt. Während DEL HOYO et al. (2006) in Fibroblastenkulturen von 13 Huntington-Patienten zwar eine signifikant erniedrigte spezifische Aktivität der für den Abbau von oxidativen Stress ebenfalls wich-tigen Katalase, nicht aber von Q10, Gluthation-Peroxidase (GPx) oder SOD fanden, wiesen CHEN
et al. (2007) eine Reduktion der Cu/Zn-SOD und der GPx in Erythrozyten nach. Die Arbeitsgruppe um F. BEAL wies 1997 post mortem im Gehirn von Huntington-Patienten eine leicht reduzierte Akti-vität von SOD im parietalen Kortex und im Zerebellum nach. Untersuchungen bei Huntington-Patienten: bisher keine veröffentlicht. Der menschliche Organismus produziert SOD im ausreichenden Maße. Eine Zufuhr durch Nah-rungsmittel ist daher bei gesunden Erwachsenen weder nötig, noch möglich, da sie im Ma-gen/Darmtrakt abgebaut wird. Durch einen Überzug mit Gliadin wird oral zugeführte SOD resor-bierbar. Ob Huntington-Kranke von einer zusätzlichen Gabe profitieren, kann beim derzeitigen Kenntnisstand wissenschaftlich nicht abschließend beurteilt werden. Ein schädlicher Einfluss ist eher unwahrscheinlich, ein Nutzen theoretisch denkbar durch den Schutz bei oxidativem Stress.
Vitamin B1 (Thiamin)
Bezug zur HK: Zitratzyklus und kataboler Stoffwechsel.
Bei der HK liegen Störungen des Polynukleotid-, Fett-, Eiweiß- und Zuckerstoffwechsels vor, die
einem latenten Typ1-Diabetes ähnlich sind, und Typ1-Diabetes tritt bei Huntington-Kranken häufi-
ger auf.
Bei Diabetikern besteht ein erhöhter Bedarf an Thiamin zur Vermeidung von diabetischen Nerven-
schädigungen. Dabei hat sich eine Behandlung mit Benfotiamin bewährt.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht.
Der normale Tagesbedarf von 1 mg Thiamin ist durch ca. 100 g Schweinefleisch gedeckt. Ob Hun-
tington-Kranke von einer Behandlung mit Benfotiamin profitieren, kann beim derzeitigen Kenntnis-
stand wissenschaftlich nicht abschließend beurteilt werden. Ein schädlicher Einfluss ist unwahr-
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
scheinlich, ein Nutzen theoretisch denkbar durch den Schutz bei einer prädiabetischen Stoffwech-sellage. Benfotiamin ist als Arzneimittel im Handel.
α-Liponsäure
Bezug zur HK: Zitratzyklus und Schutz vor oxidativem Stress.
Als Coenzym hat die α-Liponsäure enge Beziehung zum Thiamin und ist ein starkes Antioxidans,
das verbrauchte Antioxidantien wie Vitamin C und E, Coenzym Q10 oder Glutathion regeneriert. Sie
ist fett- und wasserlöslich und dringt gut ins Gehirn. Im gesunden menschlichen Organismus wird ausreichend α-Liponsäure synthetisiert. Mangelzustände können bei Lebererkrankungen, Diabetes mellitus und peripheren Neuropathien vorliegen. Die diabetische Polyneuropathie lässt sich mit α-Liponsäure 600 – 1200 mg/d wirksam behandeln. Diabetes mellitus tritt bei Huntington-Kranken gehäuft auf, oft lässt sich eine prädiabetische Stoffwechsellage nachweisen. ANDREASSEN et al. (2001) konnten bei der Huntington-Maus durch α-Liponsäure die Überlebenszeit verlängern. Neue Untersuchungen (HAGER et al., 2007) zeigten eine positive Wirkung von 600 mg α-Liponsäure bei Patienten mit Morbus Alzheimer, bei dem oxidativer Stress auch eine pathogenetische Rolle spielt. Untersuchungen bei Huntington-Patienten: keine veröffentlicht. α-Liponsäure ist nur in wenigen Lebensmitteln in größerer Menge enthalten, dazu gehören Inne-reien wie Herz und Leber sowie Seafood. 100 g Fleisch enthalten etwa 5 mg α-Liponsäure. Bisher fehlt es an Empfehlungen für den täglichen Bedarf. Ein eindeutiger Mangel an α-Liponsäure wurde bisher nicht beobachtet. Jedoch wurden niedrige Blutspiegel bei Diabetes mellitus sowie bei eini-gen neurologischen Krankheiten festgestellt. Ob Huntington-Kranke von einer Behandlung mit α-Liponsäure profitieren, kann beim derzeitigen Kenntnisstand wissenschaftlich nicht abschließend beurteilt werden. Ein schädlicher Einfluss ist unwahrscheinlich, ein Nutzen denkbar durch den Schutz vor oxidativen Stress und gegen Demenz. Bei einer prädiabetischen Stoffwechsellage soll-ten unbedingt 600 mg/d gegeben werden. α-Liponsäure ist als Arzneimittel im Handel.
Vitamin B2 (Riboflavin)
Bezug zur HK: Energie- und Proteinstoffwechsel.
Riboflavin ist im Energie- und Proteinstoffwechsel von entscheidender Bedeutung, Hinweise auf
eine gestörte Funktion oder erhöhten Bedarf bei Huntington-Kranken sind nicht publiziert.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht. Der normale Tagesbedarf von 1,2 mg ist durch ca. 40 g Schweine- oder Rinderleber bzw. 600 g Vollmilch gedeckt. Ein erhöhter Bedarf besteht bei der Langzeiteinnahme von trizyklischen Antide-pressiva oder oralen Kontrazeptiva, bei Huntington-Kranken ist ein erhöhter Bedarf wissenschaft-lich nicht belegt und nicht wahrscheinlich.
Vitamin B3 (Niacin) und Nicotinamid-Adenin-Dinucleotid (NAD)
Niacin fasst die Vitamine Nikotinsäure, Nikotinamid und die Coenzyme Nikotinamid-Adenin-
Dinukleotid (NAD) und Nikotinamid-Adenin-Dinukleotidphosphat (NADP) zusammen.
Bezug zur HK: Oxidations- und Reduktionsvorgänge, Energiestoffwechsel, Glykolyse.
Die reduzierten Coenzyme NAD(P)H tragen erheblich zum antioxidativen System der Zellen bei.
Ein Mangel an Niacin ist selten, da es aus der Aminosäure Tryptophan hergestellt werden kann,
dabei ist Vitamin B6 notwendig. Niacinmangel führt neben Hauterkrankungen zu psychiatrischen
und neurologischen Symptomen bis hin zur Niacinmangel-Enzephalopathie. Der Zusammenhang
zwischen Niacinmangel und spezifischen Läsionen im Zentralnervensystem ist bisher noch kaum
aufgeklärt.
In vielen Studien wurde die Wirkung von extern verabreichtem NADH sowie dessen Vorläufern
NAD+ und Nicotinamid auf den Organismus untersucht. In den letzten Jahrzehnten entstanden
mehrere Forschungsansätze, die von der zentralen Rolle des NADH ausgehend, dessen Einfluss
auf pathogenetische und symptomatische Gesichtspunkte verschiedener Krankheiten (mito-
chondriale Erkrankungen: Morbus Parkinson, Alzheimer; etc.) untersuchten. Die Wirkung von
NADH wurde unter anderem im Rahmen der Parkinsontherapie (BIRKMAYER et al., 1989, 1991 und
1993), der Therapie von Depressionen (BIRKMAYER et al., 1991; REX et al., 2004a) sowie in Bezug
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
auf Gedächtnisleistung und Lernfähigkeit (REX et al., 2004b, YANG et al., 2004) erforscht. Oral und parenteral verabreichtes NADH führte bei depressiven Patienten (BIRKMAYER et al., 1991) zu einer Verbesserung der Symptome um ca. 11 %. HANKES et al. (1991) fanden beim Transport von Nikotinsäure und Nikotinamid durch die Blut/Hirn-Schranke keine signifikanten Differenzen zwischen normalen Versuchspersonen und Patienten mit Huntington- oder Alzheimer-Krankheit. Untersuchungen bei Huntington-Patienten: keine veröffentlicht. Nikotinamid ist auch in hohen Dosen bis 1 g gut verträglich, während Nikotinsäure in Dosen über 30 mg vasodilatierend wirkt und zu einer Hautrötung führt. Tagesdosen von über 1 g beeinflussen den Lipoprotein- und den Kohlehydratstoffwechsel. Der normale Tagesbedarf von ca. 13 mg Niacin ist durch ca. 100 g Schweine- oder Rinderleber zu decken. Ob Huntington-Kranke von einer zusätzlichen Gabe von Niacin profitieren, kann beim der-zeitigen Kenntnisstand wissenschaftlich nicht abschließend beurteilt werden. Ein schädlicher Ein-fluss ist unwahrscheinlich, ein Nutzen denkbar bei depressiver Stimmungslage.
Vitamin B5 (Pantothensäure)
Bezug zur HK: Zitratzyklus, Auf- und Abbau von Kohlenhydraten, Fetten, Aminosäuren.
Eine Mutation der Pantothenatkinase2, die für die Synthese von Coenzym A aus der Panto-
thensäure notwendig ist, führt zu einer neurodegenerativen Erkrankung, der PKAN, früher bekannt
unter dem Begriff Morbus Hallervorden-Spatz. Hinweise auf eine gestörte Funktion oder erhöhten
Bedarf bei Huntington-Kranken sind nicht publiziert. Bei Stress wird ein höherer Bedarf vermutet.
Diabetiker scheiden eine erhöhte Menge an Pantothensäure im Urin aus und haben dadurch einen
erhöhten Bedarf.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht.
Der normale Bedarf von 6 mg Pantothensäure pro Tag ist durch ca. 100 g Schweine- oder Rinder-
leber oder Hering gedeckt. Ob Huntington-Kranke von einer zusätzlichen Gabe profitieren, kann
beim derzeitigen Kenntnisstand wissenschaftlich nicht abschließend beurteilt werden. Ein schädli-
cher Einfluss ist unwahrscheinlich, ein Nutzen ist denkbar, wenn eine erhöhte Ausscheidung im
Urin oder gleichzeitig ein Diabetes mellitus vorliegt.
Vitamin B6 (Pyridoxin)
Bezug zur HK: Aminosäuren-, Neurotransmitter- und Eiweißstoffwechsel.
Zahlreiche Untersuchungen belegen Störungen im Aminosäuren-, Neurotransmitter- und Eiweiß-
stoffwechsel bei der HK (s. oben). MANYAM et al. (1987) konnten durch die kombinierte Gabe von
Isoniazid und Pyridoxin die Konzentration von γ-Aminobuttersäure (GABA), Aspartat, Asparagin,
Homocarnosin, Ornithin, Histidin, α-Aminobuttersäure, Isoleucin, Leucin und Alanin im Liquor von
Huntington-Patienten erhöhen. Hohe Pyridoxin-Gaben können durch Steigerung der Decarboxyla-
se-Aktivität die Umwandlung von L-DOPA in Dopamin steigern, was sich schädlich auf Nervenzel-
len im Striatum auswirken könnte.
Untersuchungen bei Huntington-Patienten:
PAULSON (1971) behandelte eine Gruppe von Patienten mit HK bzw. Spätdyskinesien mit Pyridoxin
und konnte keinen positiven Effekt auf die Motorik feststellen. MCLEOD & DE L. HORNE (1972) be-
handelten 4 Huntington-Patienten für 14 Tage mit 7 g L-Tryptophan und 70 mg Pyridoxin und fan-
den auch keine Veränderung in der Motorik. BARR et al. (1978) behandelten 5 Huntington-Kranke
über 2Jahre mit 25 g L-Glutamat und 500 mg Pyridoxin, sie sahen weder in der Motorik noch im
Verhalten eine Verbesserung. STOBER et al. (1983) behandelten 11 Huntington-Patienten für 3 - 17
Monate mit hohen Dosen von Isoniazid und 3x 40 mg Pyridoxin pro Tag. Pyridoxin wurde gege-
ben, um dem negativen Einfluss des Isoniazid auf den Pyridoxin-Spiegel entgegenzuwirken. 4 Pa-
tienten wiesen eine eindeutige Besserung der Symptome und eine Abnahme der Behinderung auf,
1 Patient mit der rigiden Form (Westphal-Variante) verschlechterte sich und die restlichen 6 Pati-
enten zeigten keine Änderung des Zustandsbildes. Ob für die Wirkungen - soweit beobachtbar -
das Isoniazid oder das Pyridoxin verantwortlich waren, lässt sich aus der Untersuchung nicht
schlüssig herleiten.
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
Der für gesunde Frauen empfohlene Tagesbedarf von 1,2 mg Pyridoxin wird üblicherweise durch die Nahrung gedeckt, z. B. 120 g Lachs oder 60 g Weizenkleie. Erhöhter Bedarf besteht in der Schwangerschaft und Stillzeit sowie bei Einnahme bestimmter Medikamente, z.B. oraler Kontra-zeptiva oder L-DOPA. Kurzfristige Gabe hoher Dosen (2 bis 6 g täglich) und langfristige Gaben von mehr als 50 mg pro Tag sind neurotoxisch und führen zu einer peripheren sensorischen Neu-ropathie. Ob Huntington-Kranke von einer zusätzlichen Gabe von Pyridoxin profitieren, kann beim derzeiti-gen Kenntnisstand wissenschaftlich nicht abschließend beurteilt werden. Bei hohen Dosen ist eine neurotoxische Wirkung zu erwarten, eine langfristige Gabe von weniger als 20 mg pro Tag ist wahrscheinlich auch langfristig unbedenklich, ein positiver Einfluss auf die HK ist aber unwahr-scheinlich.
Vitamin B12 (Cobalamin)
Bezug zur HK: glutamaterge Exzitotoxizität, Mitochondrienschutz.
Cobalamin spielt eine wichtige Rolle für die Integrität der Mitochondrien. Cobalaminmangel ist mit
übermäßiger Produktion exzitotoxischer Substanzen in Zusammenhang gebracht worden. Weitere
Studien haben einen neuroprotektiven Effekt von Methylcobalamin gegen glutamaterge Exzitotoxi-
zität belegt.
BONILLA et al. (1991) fanden bei der Untersuchung von 18 Huntington-Patienten im Serum keine
Reduktion von Cobalamin oder Folsäure.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht.
Bei gesunden Frauen ist der normale tägliche Bedarf von ca. 4 µg in der Nahrung bereits durch ca.
10 g Rinderleber und 1 Hühnerei gedeckt. Die maximal aktiv resorbierbare Menge Cobalamin von
1,5 µg ist bereits bei oraler Gabe von 10 µg erreicht. Ob Huntington-Kranke von einer zusätzlichen
parenteralen Gabe von Cobalamin profitieren, kann beim derzeitigen Kenntnisstand wissenschaft-
lich nicht abschließend beurteilt werden. Ein schädlicher Einfluss ist unwahrscheinlich, ein Nutzen
ist denkbar durch Schutz vor glutamaterger Exzitotoxizität und Mitochondrien-Schutz. Ein ernied-
rigter Cobalamin-Spiegel im Serum muss auf jeden Fall korrigiert werden, was dann als kassen-
ärztliche Leistung erfolgen kann.
Folsäure (Vitamin B11)
Bezug zur HK: NMDA- und Kaininsäure-Rezeptoren, DNS-Synthese.
In einem in-vitro-Modell der HK fanden WUA et al. (2006), dass Folsäure, Gabapentin und Lamotri-
gin Neurone nicht vor durch Glutamat verursachtem Zelltod schützten, die Neurone aber durch
Memantin und Riluzol geschützt wurden. Einige Folate binden im Säugetier-Gehirn an die exzitato-
rischen Kaininsäure-Rezeptoren und scheinen eine agonistische Wirkung an diesen Rezeptoren
zu haben. Da Kaininsäure ein starkes Neurotoxin ist, ist es möglich, dass Folate diese Giftigkeit
auch besitzen und hohe Folat-Spiegel so Neurone schädigen könnten.
Eine Erhöhung von Homocystein, das als ein teilweiser Aktivator der NMDA-Rezeptoren wirkt und
so Neurone zusätzlich schädigen könnte, kann durch Substitution mit Folsäure bzw. 5-
Methyltetrahydrofolat (5-MTHF) – das die höchste biologische Aktivität aufweist – wirksam behan-
delt werden. Auch bei der positiven Wirkung einer Folsäure-Substitution auf das Auftreten von
Schlaganfällen und Demenz ist wahrscheinlich die Absenkung des Homocystein-Spiegels von Be-
deutung.
Untersuchungen bei Huntington-Patienten:
VON ALBERT (1976) berichtete über eine vergebliche Behandlung der Chorea bei Huntington-
Patienten mit Folsäure.
Die empfohlene tägliche Zufuhr von ca. 400 µg Folsäure ist mit der Nahrung schwer zu erreichen,
daher macht die Folsäure-Substitution schon für die Allgemeinbevölkerung Sinn, zumal da zusätz-
liche Folsäuregaben sowie hohe Folatspiegel im Serum mit einer besseren geistigen Leistungsfä-
higkeit assoziiert sind und sich das Risiko für Schlaganfälle sowie Demenz dadurch senken lässt.
Ein besonderer Bedarf bei Huntington-Kranken ist wissenschaftlich nicht belegt. Sollte bei der Klä-
gerin die Folatkonzentration im Serum unter 3,5 ng/ml bzw. 250 ng/ml in Erythrozyten liegen oder
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
eine genetische Mutation bei Enzymen des Folatstoffwechsels oder eine Erhöhung des Homo-cystein vorliegen, wäre eine Substitution mit 5-MTHF indiziert, wobei Dosen unter 1 mg pro Tag als sicher gelten.
Vitamin B15 (Pangamsäure = Dimethylglycin)
Bezug zur HK: Atmungskette und Antioxidans.
Natriumpanganat wird in der Art eines Coenzyms an die Cytochromoxidase gebunden und aktiviert
so den letzten Schritt in der mitochondrialen Atmungskette. Mangelzustände und -folgen sind beim
Menschen dank ausreichender Synthese nicht bekannt, daher auch keine medizinische Indikation.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht.
Eine zusätzliche Gabe bei HK lässt sich wissenschaftlich nicht begründen, ist aber vermutlich un-
schädlich, wie aus dem Gebrauch bei Sportlern oder als Anti-Aging-Präparat abzuleiten ist.
Selen
Bezug zur HK: Antiapoptotisch, Schutz vor oxidativen Schäden an der DNS u. Lipiden.
Als Bestandtteil der Glutathionperoxidase ist Selen ein wichtiges Antioxidans.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht.
Als essentielles Spurenelement muss Selen zugeführt werden. Die Deutsche Gesellschaft für Er-
nährung geht von einem täglichen Bedarf von 30 - 70 µg für Menschen ab dem 16. Lebensjahr
aus, neueste Untersuchungen errechneten einen Bedarf an 90 µg, der sich durch ca. 100 g Para-
nüsse decken lässt. Bei älteren Menschen, Rauchern, Krebskranken und Menschen mit ge-
schwächtem Immunsystem besteht möglicherweise ein erhöhter Selenbedarf. Eine zusätzliche
Gabe bei HK lässt sich wissenschaftlich nicht begründen, ist bei ausreichendem Serumspiegel um
100 µg/ml nicht erforderlich. Die Menge von bis zu 500 µg Selen pro Tag gilt bei regelmäßiger Zu-
fuhr als ungiftig. Durch einen Überschuss an Selen können u.a. periphere Neuropathien entstehen.
Kupfer
Bezug zur HK: Atmungskette, oxidativer Stress, DNS-Oxidation.
Kupfer ist Bestandteil zahlreicher Enzyme, von denen bisher 16 bekannt sind. Ein Beispiel ist die
SOD, die u.a. die Zellmembranen vor Schäden durch freie Radikale schützt und so ein wichtiges
Antioxidans ist. Kupfer trägt weiter zum Elektronentransport und damit zur Gewinnung von Energie
bei. Es ist an der Synthese von Adrenalin und Noradrenalin im Nervensystem beteiligt. Im Striatum
von Huntington-Kranken findet man post mortem eine Erhöhung von Kupfer und Eisen. Wie es
dazu kommt und welche pathogenetische Bedeutung dieser Befund hat, ist noch unklar. FOX et al.
(2007) fanden im Gehirn von Huntington-Patienten und -Mäusen Veränderungen, die dafür spre-
chen, das überschüssiges Kupfer im Gehirn das Fortschreiten der HK beschleunigt. Der Befund
wird gestützt durch die Beobachtung von NGUYEN et al. (2005), dass Clioquinol, das Kupfer- und
andere Metalle im Gewebe binden kann und gegenwärtig als Mittel gegen Morbus Alzheimer er-
probt wird, bei Huntington-Mäusen die pathologischen Veränderungen reduzierte und das
Verhalten verbesserte: sie fanden eine verminderte Huntingtin-Anhäufung, verminderte Striatum-
Atrophie, verbesserte Rotarod-Leistung (testet die Geschicklichkeit der Mäuse auf einem Drehrad),
Verminderung der Gewichtsabnahme, Normalisierung von Blutzucker und Insulin-Niveau, und
Verlängerung der Lebensspanne.
Untersuchungen bei Huntington-Patienten: keine veröffentlicht.
Der durchschnittliche Tagesbedarf von 2 mg Kupfer ist enthalten in 50 g Nüssen oder 250 g
Roggenvollkornbrot. Die Einnahme von bis zu 5 Milligramm Kupfer täglich gilt als sicher. Eine zu-
sätzliche Gabe bei der HK lässt sich wissenschaftlich nicht begründen, ein zu hoher Kupferspiegel
ist zu vermeiden, da er per se das Gehirn – insbes. die Basalganglien – schädigen und das Fort-
schreiten der HK beschleunigen kann. Allerdings muss auch ein Mangel an Kupfer vermieden
werden, da dieser zu einer Schwächung der antioxidativen Abwehr und neurologischen Störungen
führt.
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Deutsche Huntington-Hilfe e.V. Duisburg
Materialien zur Huntington-Krankheit
Als Fazit der bisherigen Untersuchungsergebnisse ist bei Huntington-Patienten und –Risiko-personen eine neuroprotektive Behandlung mit Vitamin E 200 I.E. / Tag, Vitamin C in Retard-Form 1 g / Tag, ß-Carotin 6 mg / Tag, Coenzym Q10 in Nano-Präparation 3x 100 mg / Tag, Kreatin 4x 5 g
/ Tag an 6 Tagen pro Woche und 3x 2 g Lachsöl-Kapseln mit 18 % Eicosapentaensäure (EPA) und
12 % Docosahexaensäure (DHA) sinnvoll. Die langfristige Einnahme dieser Substanzen ist in
dieser Dosierung gesundheitlich unbedenklich.
Sollte bei Patienten die Folatkonzentration im Serum unter 3,5 ng/ml bzw. 250 ng/ml in
Erythrozyten liegen oder eine Erhöhung des Homocystein vorliegen, wäre eine Substitution mit
800 µg 5-MTHF indiziert. Dabei ist eine Überdosierung unwahrscheinlich.
Sollte bei bei Patienten eine prädiabetische Stoffwechsellage vorliegen, sollten unbedingt 600 mg
α-Liponsäure pro Tag gegeben werden.
Mängel an den Vitaminen Vitamin B1, Vitamin B2, Vitamin B3, Vitamin B5, Vitamin B6, Vitamin
B12 gilt es zu vermeiden. Falls dies bei bei Patienten durch adäquate Ernährung nicht erreicht
werden kann (ggf. ist hier eine Ernährungsberatung der Familie sinnvoll), ist eine Substitution
angezeigt und kann vom Hausarzt verordnet werden.
Mängel an Kupfer, Eisen und Selen, Magnesium im Serum müssen vermieden werden, falls dies
bei bei Patienten diätetisch nicht erreicht werden kann, ist eine Substitution angezeigt, wobei
Überdosierungen unbedingt vermieden werden müssen.
Zusammenfassend ist also festzustellen:
Therapie mit Coenzym Q10, Vitamin E und Vitamin C in Kombination, ß-Carotin, Kreatin,
Eicosapentaensäure und Docosahexaensäure aussichtsreich.
Eine Folsäure-Substitution ist unabhängig von bestehenden Erkrankungen aus prophylakti-
schen Gründen schon für die Allgemeinbevölkerung sinnvoll, bei nachgewiesenem Mangel
muss substituiert werden.
Eine Substitution von Vitamin B1, Vitamin B2, Vitamin B3, Vitamin B5, Vitamin B6, Vitamin
B12, Selen und Kupfer ist nur bei nachgewiesenem Mangel angezeigt.
SOD (Superoxid-Dismutase): oral zugeführt nur wirksam, wenn mit Gliadin-Überzug (Gli-
SODin®) eingenommen. Ein Nutzen ist theoretisch denkbar durch den Schutz bei oxidati-
vem Stress. Positive Wirkungen sind bei Patienten mit kardio-vaskulären Erkrankungen be-
schrieben.
Vita Lecithin, Vit-Amino und Taurin: Substitution wissenschaftlich nicht zu begründen, auf
ausreichende Zufuhr ist zu achten und mit der Nahrung zu erreichen.
Welche konkreten Nahrungsergänzungsmittel / Vitaminpräparate kann man Huntington-
Kranken empfehlen?
Vitamin E 200 I.E. / Tag, Vitamin C in Retard-Form 1 g / Tag, ß-Carotin 6 mg / Tag, Coen-zym Q10 in Nano-Präparation (Sanomit®) 3x 100 mg / Tag, Kreatin 4x 5 g / Tag - 6x pro Woche und 3x 2 g Lachsöl-Kapseln mit 18 % Eicosapentaensäure (EPA) und 12 % Doco-sahexaensäure (DHA). Zur Folsäure-Substitution 400 µg 5-MTHF, dabei ist eine Überdosis unwahrscheinlich. Sollte bei Huntington-Kranken eine prädiabetische Stoffwechsellage vorliegen, sollte unbe-dingt α-Liponsäure 600 mg/d gegeben werden. Mängel an den Vitaminen B1, B2, B3, B5, B6 und B12 gilt es zu vermeiden; falls dies bei Patienten durch adäquate Ernährung nicht erreicht werden kann (ggf. ist hier eine Ernäh-rungsberatung sinnvoll), ist eine Substitution angezeigt und kann vom Hausarzt verordnet werden. Mängel an Kupfer, Eisen und Selen, Magnesium im Serum sind zu vermeiden, falls dies bei Patienten diätetisch nicht erreicht werden kann, ist eine Substitution angezeigt, wobei eine Überdosierung unbedingt vermieden werden muss.
Dr. Herwig Lange: Neuroprotektion (Juni 2008)
Source: http://www.huntington-hilfe.de/files/180_Lange__Neuroprotektion_2008.pdf
Alimentary Pharmacology and Therapeutics Review article: vitamin D and inflammatory bowel diseases V. P. Mouli* & A. N. Ananthakrishnan†,‡ *Department of Gastroenterology, All India Institute of Medical Sciences, New Delhi, India.† Harvard Medical School, Boston, Vitamin D is traditionally associated with bone metabolism. The immuno- ‡Division of Gastroenterology,
New clinical trial investigates APOKYN for treating debilitating morning akinesia in Park. Page 1 of 4 May 13, 2013 11:09 AM Eastern Daylight Time New clinical trial investigates APOKYN for treating debilitating morning akinesia in Parkinson's disease patients LOUISVILLE, Ky.--(BUSINESS WIRE)--US WorldMeds today announced the launch of a new clinical trial investigating APOKYN® (apomorphine hydrochloride injection) as a rapid and reliable treatment for "morning akinesia" in Parkinson's disease. AM IMPAKT, short for Apokyn for Motor IMProvement of Morning AKinesia Trial, is a Phase IV, multi-center, open-label study that will enroll approximately 100 subjects at 12 study sites across the US.



